Composición: Cada comprimido recubierto de Isentress para administración oral contiene: 434.4 mg de Raltegravir Potásico (como sal), equivalente a 400 mg de Raltegravir (Fenol libre). Excipientes: Celulosa Microcristalina, Lactosa Monohidratada, Fosfato de Calcio Dibásico Anhidro, Hipromelosa 2208, Poloxámero 407 (contiene 0.01% de Hidroxitolueno Butilato como Antioxidante), Estearil Fumarato de Sodio, Estearato de Magnesio. Además, la película de recubrimiento contiene los siguientes excipientes: Alcohol Polivinílico, Dióxido de Titanio, Polietilenglicol 3350, Talco, Oxido de Hierro Rojo y Oxido de Hierro Negro.
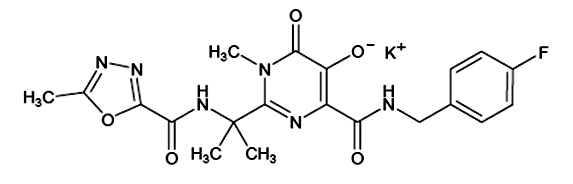
Descripción: Comprimidos recubiertos de forma oval de 400 mg, de color rosa, con "227" marcado en un lado. Isentress contiene: raltegravir potásico, un inhibidor de la transferencia de cadena de la integrasa del VIH. El nombre químico para raltegravir potásico es sal monopotásica de N-[(4-Fluorofenil) metil]-1,6-dihidro-5-hidroxi-1-metil-2-[1-metil-1-[[(5-metil-1, 3,4-oxadiazol-2-yl)carbonil]amino]etil]-6-oxo-4-pirimidinecarboxamida. La fórmula empírica es C20H20FKN6O5 y el peso molecular es 482.51. La fórmula estructural es:
Raltegravir potásico es un polvo de color blanco a blanquecino. Es soluble en agua, poco soluble en metanol, muy poco soluble en etanol y acetonitrilo e insoluble en isopropanol.
Indicaciones: Isentress está indicado, en combinación con otros agentes antirretrovirales, para el tratamiento de la infección por virus de inmunodeficiencia humana (VIH-1) en pacientes adultos. Esta indicación está basada en análisis de niveles plasmáticos de ARN de VIH-1 en 3 estudios controlados, doble ciego de hasta 48 semanas de Isentress. 2 de estos estudios fueron llevados a cabo en adultos clínicamente avanzados que ya se habían sometido a tratamiento con 3 clases de antirretrovirales (NNRTI-inhibidores de la transcriptasa inversa no análogos de nucleósidos, NRTI - inhibidores nucleósidos de la transcriptasa inversa y PI - inhibidores de la proteasa [por sus siglas en inglés]) 1 fue llevado a cabo en pacientes sin tratamiento previo. El uso de otros agentes activos con Isentress se asocia a una mayor probabilidad de respuesta al tratamiento. No se han establecido la seguridad y la eficacia de Isentress en pacientes pediátricos.
Propiedades: Mecanismo de Acción. Raltegravir es un medicamento antiviral VIH-1. Farmacodinámica: En un estudio de monoterapia, raltegravir (400 mg 2 veces al día) demostró una rápida actividad antiviral con una reducción significativa de la carga viral de 1.66 log10 copias/ml por 10 días. En el protocolo 005 y los protocolos 018 y 019, ensayos randomizados, doble ciego, controlado con placebo, dosis-respuesta, las respuestas antivirales fueron similares entre los sujetos sin importar la dosis. Efectos en el electrocardiograma: En un estudio cruzado, randomizado, placebo controlado, a 31 sujetos sanos se les administró 1 sola dosis oral supra-terapéutica de raltegravir 1600 mg y placebo. El pico de concentraciones plasmáticas de raltegravir fue aproximadamente 4 veces mayor que el pico de las concentraciones luego de una dosis de 400 mg. Isentress no parece prolongar el intervalo QTc durante 12 horas post dosis. Después de ajustar la línea base y placebo, el cambio en la media máxima QTc fue -0.4 mseg (IC del 95% 1 cola > 3.1 mseg). Farmacocinética: Absorción: Raltegravir es absorbido con una Tmáx post dosis de aproximadamente 3 horas en ayunas. El AUC y Cmáx de la dosis de raltegravir aumenta proporcionalmente sobre el rango de dosis de 100 mg a 1600 mg. La C12hr de la dosis de raltegravir aumentó proporcionalmente la dosis sobre el rango de dosis de 100 a 800 mg y aumentó ligeramente menos que proporcionalmente la dosis sobre el rango de dosis de 100 mg a 1600 mg. Con una dosis de 2 veces al día, el estado de equilibrio es alcanzado aproximadamente dentro de los 2 primeros días de administrada la dosis. Hubo una leve a nula acumulación en el AUC y Cmax. El rango promedio de acumulación para C12hr varió en aproximadamente 1.2 a 1.6. La biodisponibilidad absoluta de raltegravir no ha sido establecida. En sujetos que recibieron 400 mg solo 2 veces al día, las exposiciones al medicamento raltegravir fueron caracterizadas por una media geométrica AUC0-12hr de 14.3 µM•hr y C12hr de 142 nM. Se observó una considerable variabilidad en la farmacocinética de raltegravir. Para lo observado en los Protocolos 018 y 019 para C12hr, el coeficiente de variación (CV) para la variabilidad inter-sujeto = 212% y el CV para la variabilidad intra-sujeto = 122%. Efecto de los alimentos en la absorción oral: Isentress puede ser administrado con o sin alimentos. Raltegravir se administró sin considerar la alimentación como crucial en estudios de seguridad y de eficacia en pacientes infectados por VIH-1. Los efectos del consumo de comida baja, moderada y alta en grasa en la farmacocinética del raltegravir en estado de equilibrio fue evaluado en voluntarios sanos. La administración de múltiples dosis de raltegravir después de una comida moderada en grasa (600 Kcal, 21 g grasa) no afectó el AUC de raltegravir en un grado clínicamente significativo con un incremento de 13% con respecto al ayuno. La C12hr de raltegravir fue 66% superior y la Cmáx fue 5% superior después de una comida moderadamente grasa con respecto al ayuno. La administración de raltegravir después de una comida alta en grasas (825 Kcal, 52 g grasa) aumentó el AUC y la Cmáx aproximadamente 2 veces y aumentó el C12hr 4.1 veces. La administración de raltegravir después de una comida baja en grasas (300 Kcal, 2.5 g grasa) disminuyó el AUC y la Cmáx en 46% y 52%, respectivamente; C12hr no tuvo cambios. La alimentación parece aumentar la variabilidad farmacocinética en relación con el ayuno. Distribución: Raltegravir se une aproximadamente en un 83% a las proteínas plasmáticas humanas sobre el rango de concentración de 2 a 10 µM. Metabolismo y excreción: La vida media terminal aparente de raltegravir es de aproximadamente 9 horas, con una vida media fase α más corta (~1 hora) contando para la mayor parte del AUC. Luego de la administración de una dosis oral radiomarcada de raltegravir, un 51 y 32% de la dosis aproximadamente fue excretada en las heces y la orina, respectivamente. En las heces, sólo raltegravir estuvo presente, la mayoría de lo cual probablemente derive de la hidrólisis de raltegravir glucuronido secretado en la bilis como se observa en las especies preclínicas. 2 componentes, llamados raltegravir y raltegravir glucurónido, fueron detectados en la orina y representan el 9 y 23% aproximadamente de la dosis, respectivamente. La principal entidad circulante fue raltegravir y representó un 70% aproximadamente de la radioactividad total; la radioactividad restante en el plasma fue representada por raltegravir glucurónido. Estudios que usaron inhibidores químicos isoforma selectivos y el cADN expresado UDP-glucuronosiltransferasas (UGT) muestran que la UGT1A1 es la principal enzima responsable de la formación de raltegravir glucurónido. De este modo, los datos indican que el principal mecanismo de depuración de raltegravir en humanos es la glucuronidación mediada por UGT1A1. Poblaciones especiales: Pediátricos: No se ha establecido la farmacocinética de raltegravir en pacientes pediátricos. Edad: Los efectos de la edad en la farmacocinética de raltegravir fueron evaluados en un análisis compuesto. No es necesario un ajuste de la dosis. Raza: Los efectos de la raza en la farmacocinética de raltegravir fueron evaluados en un análisis compuesto. No es necesario un ajuste de la dosis. Género: Se desarrolló un estudio de la farmacocinética de raltegravir en mujeres y hombres adultos sanos. Adicionalmente, el efecto del género fue evaluado en un análisis compuesto de datos de la farmacocinética de 103 sujetos sanos y 28 sujetos infectados con VIH-1 que recibieron monoterapia con raltegravir administrado en ayunas. No es necesario un ajuste de la dosis. Insuficiencia hepática: Raltegravir es eliminado principalmente por la glucuronidación en el hígado. Un estudio en la farmacocinética de raltegravir se desarrolló en sujetos con una insuficiencia hepática moderada. Adicionalmente, la insuficiencia hepática fue evaluada en un análisis farmacocinético compuesto. No hubo diferencias farmacocinéticas clínicamente importantes entre sujetos con insuficiencia hepática moderada y sujetos sanos. No es necesario un ajuste en la dosis para pacientes con insuficiencia hepática media a moderada. No ha sido estudiado el efecto en la farmacocinética de raltegravir en pacientes con insuficiencia hepática severa. Insuficiencia renal: La depuración renal del medicamento inalterado es una menor vía de eliminación. Un estudio en la farmacocinética de raltegravir se desarrolló en sujetos con insuficiencia renal severa. Adicionalmente, la insuficiencia renal fue evaluada en un análisis farmacocinético compuesto. No hubo diferencias farmacocinéticas clínicamente importantes entre sujetos con insuficiencia renal severa y sujetos sanos. No se requiere un ajuste de la dosis. Debido a que se desconoce el grado en que Isentress puede ser dializable, se debe evitar una dosis antes de una sesión de diálisis. Polimorfismo UGT1A1: No hay evidencia de que el polimorfismo común UGT1A1 altere la farmacocinética de raltegravir de forma clínicamente significativa. En una comparación de 30 sujetos con genotipo *28/*28 (asociado con reducida actividad de UGT1A1) a 27 sujetos con genotipo de estado salvaje, la media geométrica (90% CI) de AUC fue 1.41 (0.96, 2.09). Interacciones con otros medicamentos:
Microbiología: Mecanismo de acción: Raltegravir inhibe la actividad catalítica del VIH-1 integrasa, que codifica la enzima que es requerida para la replicación viral. La inhibición de la integrasa previene la inserción covalente, o integración, del ADN VIH-1 lineal no integrado dentro del genoma de células huésped previniendo la formación del provirus VIH-1. El provirus se requiere para dirigir la producción del virus progenie, por lo que la inhibición de la integración previene la propagación de la infección viral. Raltegravir no inhibe significativamente las fosforiltransferasas humanas incluyendo las ADN polimerasas α, ß y Y. Actividad antiviral en el cultivo celular: Raltegravir a concentraciones de 31 ± 20 nM resultó en un 95% de inhibición (EC95) de propagación viral (relativo a cultivos no tratados de virus infectados) en humanos de cultivos de células T-linfoide infectado con líneas celulares adaptadas de VIH-1 variante H9IIIB. Además, 5 aislamientos clínicos de VIH-1 del subtipo B tenían valores de EC95 que van desde 9 a 19 nM de células mononucleares cultivadas de sangre periférica humana mitógena-activada. En un único ensayo de ciclo de infección, el raltegravir inhibe la infección de 23 VIH-1 aislados que representan 5 subtipos no B (A, C, D, F y G) con valores EC50 que van desde 5 a 12 nM. Raltegravir además inhibió la replicación de un aislado de VIH-2 cuando éste se probó en células CEMx174 (valor EC95 = 6 nM). Adicionalmente a una actividad antirretroviral sinérgica que se observó cuando las células T linfoides infectadas con la variante del VIH-1 H9IIIB fueron incubadas con raltegravir en combinación con inhibidores de la transcriptasa reversa no-nucleósidos (delavirdina, efavirenz o nevirapina); inhibidores de la transcriptasa reversa análogos de nucleósidos (abacavir, didanosina, lamivudina, estavudina, tenofovir, zalcitabina o zidovudina); inhibidores de proteasa (amprenavir, atazanavir, indinavir, lopinavir, nelfinavir, ritonavir o saquinavir); o el inhibidor de entrada enfuvirtida. Resistencia: Las mutaciones observadas en la secuencia de codificación de la integrasa del VIH-1 que contribuyeron a la resistencia de raltegravir (evolución ya sea en cultivos celulares o en pacientes tratados con raltegravir) generalmente incluyeron una sustitución de aminoácidos ya sea en Q148 (que cambió a H, K, o R) o en N155 (cambió a H) más 1 o más sustituciones adicionales (por ej. L74M, E92Q, T97A, E138A/K, G140A/S, V151I, G163R, H183P, Y226C/D/F/H, S230R y D232N). Las sustituciones de aminoácidos en Y/143C/H/R es otra vía para la resistencia de raltegravir. Sujetos sin tratamiento previo: A la semana 48 en el estudio STARTMRK, las sustituciones primarias asociadas a resistencia a raltegravir se observaron en 3 (1 con Y143R, y 2 con Q148H/R) de los 6 sujetos con fracaso virológico que tenían pares evaluables de datos genotípicos. Sujetos expuestos a tratamiento previo: A la semana 48 en los estudios BENCHMRK, por lo menos 1 de las 3 sustituciones primarias asociadas a resistencia a raltegravir -Y143C/H/R, Q148H/K/R, y N155H- se observó en 63 (64.3%) de los 98 sujetos con fracaso virológico que tenían datos genotípicos evaluables de pares de muestras basales y de fracaso del tratamiento con raltegravir. Algunas (n = 18) de las cepas víricas aisladas de VIH-1 que exhibieron 1 o más de las 3 sustituciones primarias asociadas a resistencia a raltegravir se evaluaron para determinar la susceptibilidad a raltegravir, y arrojaron una mediana de disminución de 47.3 veces (disminución media de 73.1 ± 60.8 veces, con un intervalo de 0.9 a 200 veces) en comparación con las cepas basales.
Posología: Para el tratamiento de pacientes con infección por VIH-1, la dosis de Isentress es de 400 mg administrados oralmente, 2 veces al día con o sin alimentos. Durante la co-administración con rifampicina, la dosis recomendada de Isentress es de 800 mg 2 veces al día con o sin alimentos.
Modo de Empleo:Uso en poblaciones específicas: Embarazo, categoría C: Se debe usar Isentress durante el embarazo sólo cuando el beneficio potencial justifique el riesgo potencial para el feto. No hay estudios adecuados y bien controlados en mujeres embarazadas. Además, no hubo estudios farmacocinéticos realizados en pacientes embarazadas. Fueron llevados a cabo estudios de toxicidad de desarrollo en conejos (con dosis orales de hasta 1000 mg/kg/día) y ratas (con dosis orales de hasta 600 mg/kg/día). El estudio de toxicidad reproductiva en ratas fue llevado a cabo con evaluación pre, peri y postnatal. Las dosis más altas en estos estudios produjeron exposiciones sistémicas en estas especies, aproximadamente de 3 a 4 veces superiores de la dosis recomendada para humanos. Tanto en conejos como en ratas, no se observaron efectos relacionados con el tratamiento sobre la supervivencia embrionaria/fetal o el peso fetal. Además, en los conejos no se observaron cambios externos, viscerales o en el esqueleto relacionados con el tratamiento. Sin embargo, hubo aumento en la incidencia de costillas supernumerarias entre las ratas tratadas con dosis de 600 mg/kg/día (una exposición de aproximadamente 3 veces superior a la dosis recomendada para los humanos). La transferencia placentaria del fármaco fue demostrada tanto en los conejos como en ratas. Con una dosis maternal de 600 mg/kg/día en ratas, las concentraciones media del fármaco en el plasma fetal fueron de aproximadamente 1.5 a 2.5 veces superiores que en el plasma materno a 1 hora y 24 horas post-dosis respectivamente. Las concentraciones media del fármaco en el plasma fetal fueron de aproximadamente el 2% de la concentración media maternal a 1 hora y 24 horas post-dosis con una dosis maternal de 1000 mg/kg/día en los conejos. Lactancia: No se aconseja la lactancia durante el uso de Isentress. Además, se recomienda que las madres infectadas por VIH no amamanten a sus hijos para evitar el riesgo de trasmisión postnatal del VIH. No se sabe si raltegravir es secretado en la leche humana. Sin embargo, raltegravir es excretado en la leche de las ratas lactantes. Las concentraciones medias del fármaco en la leche fueron aproximadamente 3 veces superiores a las del plasma materno, con una dosis materna de 600 mg/kg/día en ratas. No hubo efectos en las crías de las ratas que se atribuyeron a la exposición de Isentress a través de la leche. Uso pediátrico: No se han establecido la seguridad y la eficacia de Isentress en los pacientes pediátricos. Uso geriátrico: Los estudios clínicos de Isentress no incluyeron una cantidad suficiente de sujetos de 65 ańos o mayores para poder determinar si responden de forma distinta que los sujetos más jóvenes. No se han identificado diferencias en las respuestas entre jóvenes y personas mayores en otras experiencias clínicas reportadas. Por lo general, la dosis para un sujeto mayor debería ser elegida con cautela, debido a la mayor frecuencia de disminución en la función hepática, renal o cardíaca, y de enfermedades concomitantes u otra terapia. Uso en pacientes con insuficiencia hepática: No se observaron diferencias farmacocinéticas importantes entre los sujetos con insuficiencia hepática moderada y los sujetos sanos. No es necesario ajustar la dosis para los pacientes con insuficiencia hepática de leve a moderada. No se ha estudiado el efecto de la insuficiencia hepática severa sobre la farmacocinética de raltegravir (ver Farmacología clínica). Uso en pacientes con insuficiencia renal: No se observaron diferencias farmacocinéticas importantes entre los sujetos con insuficiencia renal severa y los sujetos sanos. No es necesario ajustar la dosis (ver Farmacología clínica).
Efectos Colaterales:Experiencia de ensayos clínicos: Debido a que los ensayos clínicos son llevados a cabo en condiciones muy variables, las tasas de reacciones adversas observadas en los ensayos clínicos de un fármaco no pueden ser comparadas directamente con las tasas de los ensayos clínicos de otro fármaco y pueden no reflejar las tasas observadas en la práctica. Estudios en pacientes sin tratamiento previo: La siguiente evaluación de la seguridad de Isentress en sujetos que no han sido expuestos a tratamiento previo se basa en el estudio aleatorizado, doble ciego y controlado con comparador activo conducido en sujetos sin tratamiento previo, Startmrk (Protocolo 021), en el que se administró Isentress 400 mg 2 veces al día en combinación con una dosis fija de emtricitabina 200 mg (+) tenofovir 300 mg (N=281), versus efavirenz (EFV) 600 mg al acostarse más emtricitabina (+) tenofovir (N=282). Durante el tratamiento doble ciego, el seguimiento total a los sujetos que recibieron Isentress 400 mg 2 veces al día + emtricitabina (+) tenofovir fue 247 paciente-ańos; y 241 ańos paciente en el caso de sujetos tratados con efavirenz 600 mg al acostarse + emtricitabina (+) tenofovir. En el Protocolo 021, la tasa de suspensiones del tratamiento debido a reacciones adversas fue 3% en los sujetos que recibieron Isentress + emtricitabina (+) tenofovir, y 6% en quienes recibieron efavirenz + emtricitabina (+) tenofovir. Las reacciones clínicas adversas relacionadas con el fármaco (ADRs, por sus siglas en inglés) listadas más abajo fueron consideradas por los investigadores causalmente relacionadas con la administración de Isentress + emtricitabina (+) tenofovir o efavirenz + emtricitabina (+) tenofovir. En la opinión de los investigadores, las reacciones clínicas adversas al fármaco (ADRs por sus siglas en inglés) listadas a continuación tuvieron una relación causal con la administración de Isentress + emtricitabina (+) tenofovir, o de efavirenz + emtricitabina (+) tenofovir. Las ADRs clínicas de intensidad moderada a severa presentadas en ≥ 2% de los sujetos sin tratamiento previo tratados con Isentress y con mayor incidencia que en quienes recibieron efavirenz se presentan en la Tabla 1.
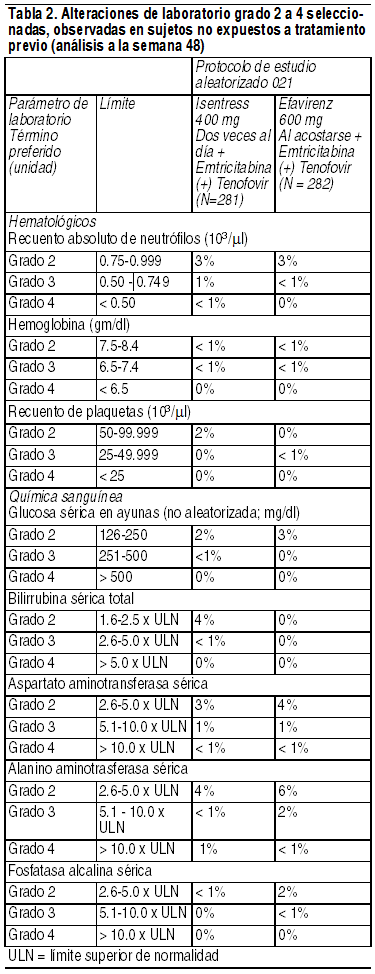
Reacciones adversas menos frecuentes: Las siguientes ADR se observaron en < 2% de los sujetos que recibieron Isentress + emtricitabina (+) tenofovir. Estos eventos se han incluido debido a su seriedad, ya que se presentan con mayor frecuencia en quienes usan Isentress al compararlos con quienes reciben efavirenz, o a que el investigador considera que existe una posible relación causal con el fármaco. Trastornos generales y condiciones en el sitio de administración: fatiga. Trastornos psiquiátricos: sueńos anormales. Alteraciones en parámetros de laboratorio: Los porcentajes de sujetos adultos tratados con Isentress 400 mg 2 veces al día o con efavirenz en el Protocolo 021, y que presentaron alteraciones de laboratorio grados 2 a 4 que representan un deterioro respecto de la condición basal se presentan en la Tabla 2.
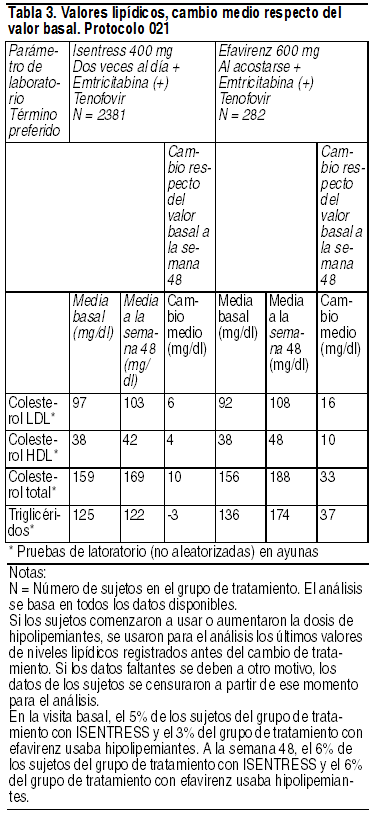
Lípidos; cambio respecto del valor basal: Los cambios de los niveles lipídicos en ayunas respecto del valor basal se muestran en la Tabla 3.
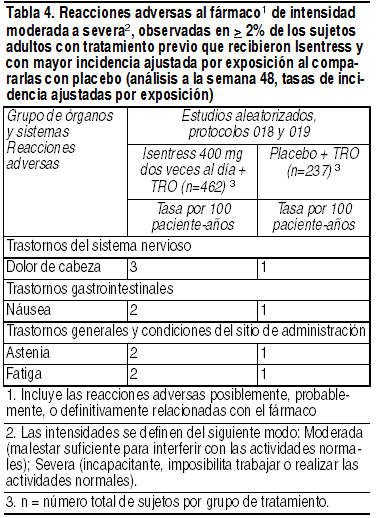
Estudios en pacientes expuestos a tratamiento previo: La evaluación de la seguridad de Isentress en sujetos expuestos a tratamiento previo se basa en los datos de seguridad agrupados obtenidos en los estudios aleatorizados, doble ciego y controlados con placebo BENCHMRK 1 y BENCHMRK 2 (protocolos 018 y 019), conducidos en sujetos adultos con infección por VIH-1 que habían recibido tratamiento previo con antirretrovirales. Un total de 462 sujetos recibió la dosis recomendada de Isentress 400 mg 2 veces al día en combinación con terapia de respaldo optimizada (TRO), comparados con 237 sujetos que recibieron placebo combinado con TRO. La mediana de duración de la terapia en estos estudios fue 48 semanas para los sujetos tratados con Isentress, y 38 semanas en los sujetos que recibieron el placebo. La exposición total a Isentress fue 387 paciente-ańos frente a 156 paciente-ańos en el caso del placebo. Las tasas de descontinuación del tratamiento debido a eventos adversos fueron 2% en los sujetos tratados con Isentress, y 3% en los que recibieron el placebo. Las ADR clínicas fueron consideradas por los investigadores causalmente relacionadas con la administración de Isentress + TRO o de placebo + TRO. Las reacciones adversas al fármaco de intensidad moderada a severa observadas en ≥ 2% de los sujetos tratados con Isentress, y que ocurrieron con mayor incidencia ajustada por exposición en comparación con el placebo, se presentan en la Tabla 4.
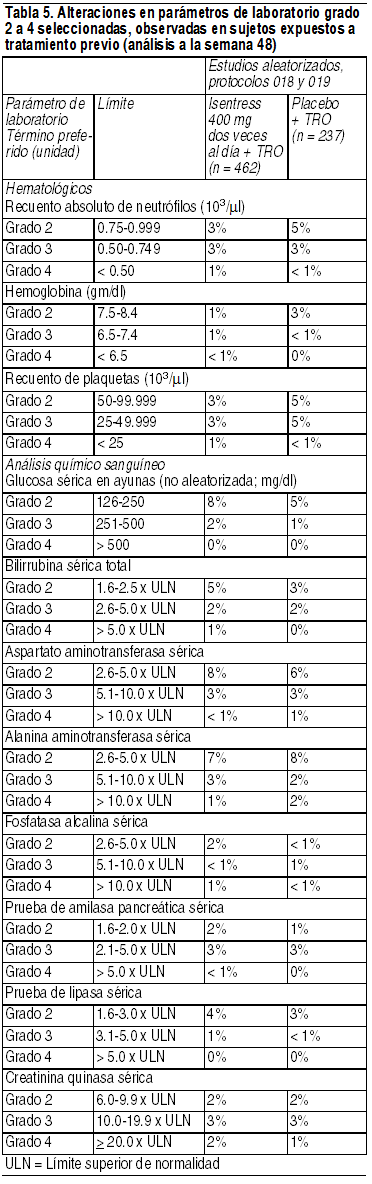
Reacciones adversas menos frecuentes: Las siguientes ADR se observaron en < 2% de los sujetos que recibieron Isentress + TRO. Estos eventos se han incluido debido a su seriedad, a que se presentaron con mayor frecuencia en quienes usaron Isentress al compararlos con quienes recibieron el placebo, o a que el investigador consideró que existe una posible relación causal con los fármacos. Trastornos gastrointestinales: Dolor abdominal, gastritis. Trastornos hepatobiliares: Hepatitis. Trastornos del sistema inmunológico: Hipersensibilidad. Infecciones e infestaciones: Herpes genital, herpes zoster. Trastornos del sistema nervioso: Mareos. Trastornos renales y urinarios: Insuficiencia renal. Alteraciones en parámetros de laboratorio: Los porcentajes de sujetos adultos tratados con Isentress 400 mg 2 veces al día o con placebo los protocolos 018 y 019, y que presentaron alteraciones en parámetros de laboratorio grados 2 a 4 que representan un deterioro respecto de la condición inicial, se presentan en la Tabla 5.
Eventos adversos seleccionados: Independientemente de su relación con el medicamento: Se reportaron cánceres en sujetos con exposición a tratamiento previo que comenzaron a recibir Isentress o placebo, ambos con TRO; y también en sujetos sin tratamiento previo que empezaron a recibir Isentress o efavirenz, ambos con emtricitabina (+) tenofovir. Varios de los casos fueron recurrentes. Los tipos y las tasas de cánceres específicos fueron los previstos en poblaciones altamente inmunodeprimidas (muchos tenían un recuento de CD4+ inferior a 50 células/mm3, y la mayoría tenía diagnóstico previo de SIDA). El grupo que recibió Isentress y el grupo que recibió el comparador exhibieron un riesgo similar de desarrollar cáncer en estos estudios. Se observaron alteraciones grados 2 a 4 en los análisis de creatina quinasa de sujetos tratados con Isentress (consulte la Tabla 5). Se han reportado casos de miopatía y rabdomiólisis; no obstante, se desconoce cuál es la relación entre Isentress y tales eventos. El medicamento debe usarse con precaución en pacientes que tienen riesgo aumentado de miopatía o rabdomiólisis, como aquellos que reciben medicación concomitante que se sabe causa tales condiciones. Rash ha ocurrido más comúnmente en sujetos con tratamiento previo recibiendo regímenes que contienen Isentress + danuravir/ritonavir comparado con sujetos que recibieron Isentress sin danuravir/ritonavir o danuravir/ritonavir sin Isentress. Sin embargo, el rash considerado relacionado al fármaco ocurrió a tasas similares para los 3 grupos. El rash fue leve a moderado en severidad y no limitó la terapia; no se documentó discontinuaciones debido a este efecto adverso. Pacientes con condiciones coexistentes: Pacientes coinfectados con el virus de la hepatitis B y/o hepatitis C: En los estudios clínicos aleatorizados, doble ciego y controlados con placebo, se permitió la inclusión de sujetos con exposición a tratamiento previo (N = 114/699 ó 16%) y de sujetos sin tratamiento previo (N = 34/563 ó 6%) que presentaran coinfección crónica (pero no aguda) activa con el virus de la hepatitis B y/o hepatitis C, siempre que los resultados de sus pruebas de función hepática basales no sobrepasaran 5 veces el límite superior de normalidad (ULN). Por lo general, el perfil de seguridad de Isentress en sujetos coinfectados con el virus de la hepatitis B y/o la hepatitis C fue similar al observado en sujetos sin coinfección con tales virus, aunque las tasas de alteraciones en los valores de AST y ALT fueron más altas en el subgrupo coinfectado con el virus de hepatitis B y/o hepatitis C en todos los grupos de tratamiento. Del grupo de sujetos con exposición a tratamiento previo tratados con Isentress, el 25%, 31% y 12% del subgrupo de sujetos coinfectados presentó alteraciones grado 2 o superiores en los niveles de AST, ALT o bilirrubina total, respectivamente, que reflejaron un deterioro respecto del grado basal; lo que se compara con el 8%, 7% y 8% observado en todos los otros sujetos tratados con Isentress. Del grupo de sujetos sin tratamiento previo tratados con Isentress, el 17%, 22% y 11% del subgrupo de sujetos coinfectados presentó alteraciones grado 2 o superiores en los niveles de AST, ALT o bilirrubina total, respectivamente, que reflejaron un deterioro respecto del grado basal; lo que se compara con el 4%, 4% y 3% observado en todos los otros sujetos tratados con Isentress. Experiencia post-comercialización: Se han identificado las siguientes reacciones adversas durante el uso de Isentress después de su aprobación. Dado que estas reacciones se notifican voluntariamente y provienen de una población de tamańo indeterminado, no siempre es posible hacer una estimación confiable de su frecuencia ni establecer una relación causal con la exposición al fármaco. Trastornos de la sangre y del sistema linfático: Trombocitopenia. Trastornos psiquiátricos: Ansiedad, depresión (especialmente en pacientes con antecedentes de enfermedad psiquiátrica), incluidas ideación y conductas suicidas, paranoia. Trastornos cutáneos y del tejido subcutáneo: Exantema, síndrome de Stevens-Johnson. Trastornos músculo-esqueléticos y del tejido conectivo: Rabdomiolisis.
Contraindicaciones: Ninguna.
Advertencias:Síndrome de reconstitución inmune: Durante la fase inicial del tratamiento, los pacientes que responden a la terapia antirretroviral pueden desarrollar una respuesta inflamatoria debida a infecciones oportunistas residuales o indolentes (tal como complejo Microbacterium avium, complejo citomegalo-virus, neumonía por Pneumocystis jiroveci, tuberculosis por micobacterium, o reactivación del virus varicela-zoster), que pueden necesitar evaluación y tratamiento adicionales.
Precauciones:Toxicología no clínica: Carcinogenésis, mutagénesis, alteraciones de la fertilidad: Los estudios de carcinogenicidad de raltegravir en ratones no mostró ningún potencial carcinogénico. En los niveles más altos de dosis, 400 mg/kg/día en hembras y 250 mg/kg/día en machos, la exposición sistémica fue de 1.8 vez (hembras) o 1.2 vez (machos) más que el AUC (54 µM•hr) en dosis de 400 mg 2 veces al día en humanos. Carcinoma de células escamosas de la nariz/nasofaringe relacionados con el tratamiento se observó en ratas hembras a dosis de 600 mg/kg/día de raltegravir durante 104 semanas. Posiblemente estos tumores sean el resultado de la irritación local y la inflamación debido a la deposición local y/o la aspiración de la droga en la mucosa de la nariz/nasofaringe durante la dosificación. No se observaron tumores de nariz/nasofaringe en ratas con dosis de 150 mg/kg/día (machos) y 50 mg/kg/día (hembras) y la exposición sistémica en ratas fue 1.7 vez (machos) a 1.4 vez (hembras) mayor que la AUC (54 µM•hr) en dosis de 400 mg 2 veces al día en humanos. Ninguna evidencia de mutagenicidad o genotoxicidad se observó en las pruebas de mutagénesis microbial in vitro (Ames), ensayos de elución in vitro para rotura de ADN y estudios de aberración cromosomal in vivo. Ningún efecto en la fertilidad fue visto en ratas hembras y machos con dosis de hasta 600 mg/kg/diarios los cuales resultan en unas 3 veces sobre la dosis de exposición humana recomendada.
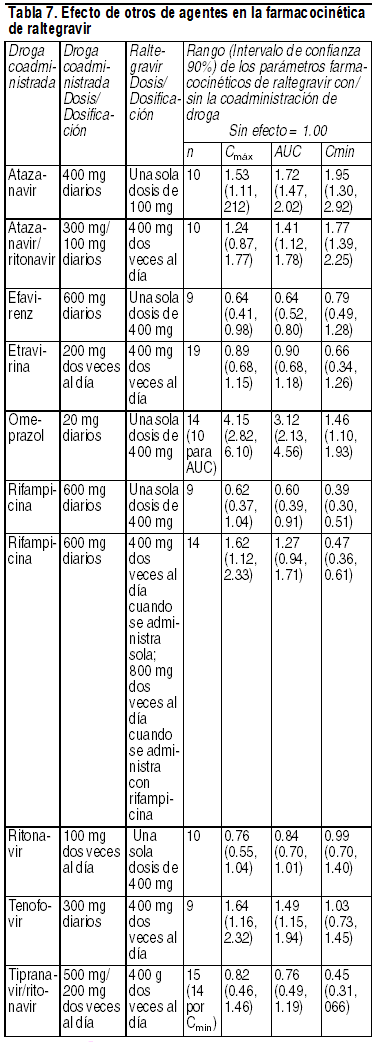
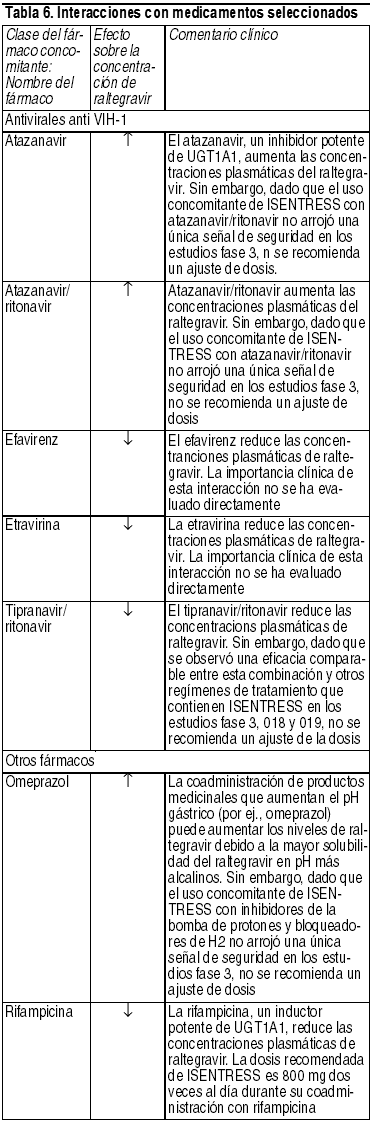
Interacciones Medicamentosas:Efecto de raltegravir en la farmacocinética de otros agentes: Raltegravir no inhibe (IC50 > 100 µM) a CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 o CYP3A in vitro. Además, raltegravir in vitro, no indujo a CYP1A2, CYP2B6 o CYP3A4. Un estudio de interacción de medicamento de midazolam confirmó la baja propensión de raltegravir para alterar la farmacocinética de agentes metabolizados por CYP3A4 in vivo, demostrando ello una falta de efecto de raltegravir en la farmacocinética de midazolam, un sustrato sensible del CYP3A4. De manera similar, raltegravir no es un inhibidor (IC50 > 50 µM) de las UDP-glucuronosiltransferasas (UGT) probadas (UGT1A1, UGT2B7), y raltegravir no inhibe el trasporte mediado por la glicoproteína P. Sobre la base de estos datos, no se espera que Isentress afecte la farmacocinética de fármacos que son substratos de estas enzimas o de la glicoproteína P (ej., inhibidores de la proteasa, NNRTI, analgésicos opioides, estatinas, antimicóticos azoles, inhibidores de la bomba de protones y medicamentos contra la disfunción eréctil). En estudios de interacción de medicamento, raltegravir no tuvo un efecto de importancia clínica sobre la farmacocinética de los siguientes: anticonceptivos hormonales, metadona, lamivudina, tenofovir, etravirina. Efecto de otros agentes sobre la farmacocinética de raltegravir: Raltegravir no es un sustrato de las enzimas del citocromo P450 (CYP). Sobre la base de estudios in vivo e in vitro, raltregravir es eliminado principalmente por vía metabólica mediante glucuronidación mediada por UGT1A1. Rifampicina, un potente inductor de UGT1A1, reduce las concentraciones plasmáticas de Isentress. Por eso, la dosis de Isentress debe incrementarse durante la co-administración (ver Posología) con rifampicina. No se conoce el impacto de otros inductores de enzimas metabolizadoras de fármaco, tal como fenitoína y fenobarbital, sobre UGT1A1. La coadministración de Isentress con otros fármacos que inhiben la UGT1A1 puede aumentar los niveles plasmáticos de raltegravir. Interacciones con medicamentos seleccionados son presentadas en la tabla 6.
Sobredosificación: No hay ninguna información específica sobre el tratamiento de la sobredosis de Isentress. Se estudiaron dosis tan altas como 1600 mg en dosis única y dosis múltiples de 800 mg 2 veces al día en voluntarios sanos sin evidencia de toxicidad. Dosis ocasionales de hasta 1800 mg por día fueron tomadas en los estudios clínicos con pacientes infectados por VIH 1, sin ninguna evidencia de toxicidad. En el caso de una sobredosis, es aconsejable emplear las medidas de apoyo estándar, por ejemplo, remover el material no absorbido del tracto gastrointestinal, monitoreo clínico (incluyendo la toma de un electrocardiograma) y establecer una terapia de soporte si es necesario. No se conoce hasta qué punto Isentress sea dializable.
 Laboratorio
Laboratorio