Composición: Cada comprimido recubierto contiene: 10 mg de Rivaroxabán. Cada comprimido recubierto de 10 mg contiene: 27.90 mg de Lactosa Monohidrato (= 26.51 mg de Lactosa) por comprimido.
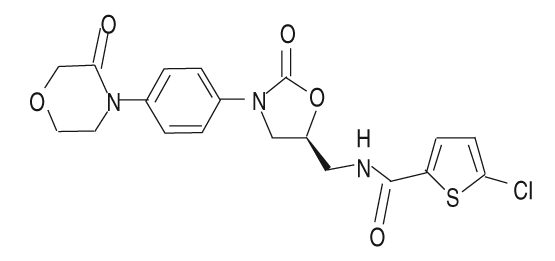
Descripción: Comprimidos recubiertos redondos, biconvexos, de color rojo claro, de 6 mm de diámetro, para uso por vía oral. La cruz de Bayer en una cara, y 10 y un triángulo en otra cara. Rivaroxabán es un enantiómero S puro. Es un polvo inodoro, no higroscópico, de color blanco a amarillento. Nombre químico: 5-cloro-N-({(5S)-2-oxo-3-[4-(3-oxo-4-morfolinil)fenil]-1,3 oxazolidin-5 il}metil)-2-tiofeno-carboxamida. Fórmula empírica: C19 H18 Cl N3 O5 S. Peso molecular: 435.89
Indicaciones: Xarelto está indicado para la prevención de la tromboembolia venosa en pacientes sometidos a una intervención quirúrgica ortopédica mayor de las extremidades inferiores.
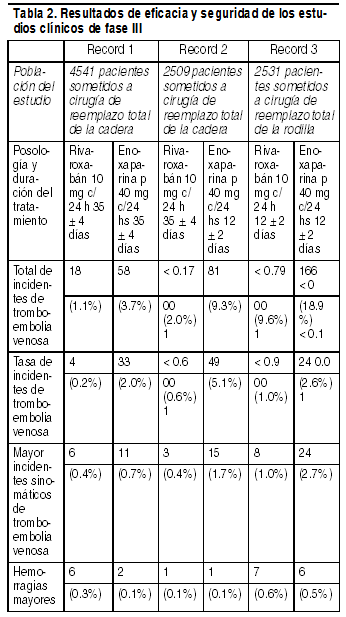
Propiedades:Propiedades farmacodinámicas: Mecanismo de acción: El rivaroxabán es un inhibidor directo del factor Xa, altamente selectivo, con biodisponibilidad oral. La activación del factor X para convertirse en factor Xa (FXa) por medio de las vías intrínseca y extrínseca desempeńa una función fundamental en la cascada de la coagulación de la sangre. El FXa convierte directamente la protrombina en trombina por medio del complejo protrombinasa y, en última instancia, esta reacción lleva a la formación de coágulos de fibrina y a la activación de plaquetas por la trombina. Una molécula de FXa puede generar más de 1000 moléculas de trombina debido a la naturaleza de amplificación de la cascada de coagulación. Además, la velocidad de reacción del FXa unido a la protrombinasa aumenta en 300.000 veces, en comparación con la del FXa libre, y causa una explosión generadora de trombina. Los inhibidores selectivos del FXa pueden eliminar la explosión amplificada generadora de trombina. En consecuencia, varias pruebas específicas y globales de coagulación son afectadas por el rivaroxabán. En seres humanos, se ha observado la inhibición dependiente de la dosis de la actividad del factor Xa. Efectos farmacodinámicos: En humanos, se observó inhibición dependiente de la dosis de la actividad del factor Xa. El tiempo de protrombina (TP) es influenciado por el rivaroxabán de manera dependiente de la dosis, con una correlación estrecha con las concentraciones plasmáticas (el valor de r es igual a 0.98) si se emplea Neoplastin para el análisis. Otros reactivos proporcionarían resultados diferentes. La lectura del TP debe hacerse en segundos, porque el INR (Indice Internacional Normalizado) sólo se ha calibrado y validado para cumarinas y no puede utilizarse para ningún otro anticoagulante. En los pacientes sometidos a una intervención de cirugía ortopédica mayor, los percentiles 5/95 del TP (Neoplastin) 2 a 4 horas después de tomar el comprimido (es decir, en el momento del efecto máximo), variaban entre 13 y 25 segundos. En un estudio de farmacología clínica sobre la reversión de la farmacodinámica de rivaroxaban en sujetos adultos sanos (n=22), se evaluaron los efectos de dosis únicas (50 UI/kg) de 2 tipos diferentes de PCC, un PCC de 3 factores (Factores II, IX, y X) y un PCC de 4 factores (Factores II, VII, IX y X). El PCC de 3 factores redujo la media de los valores del TP de Neoplastin en aproximadamente 1.0 segundo dentro de los 30 minutos, en comparación con reducciones de aproximadamente 3.5 segundos observadas con el PCC de 4 factores. En cambio, el PCC de 3 factores tuvo un efecto global más rápido y mayor en la reversión de los cambios en la generación de trombina endógena respecto del PCC de 4 factores (ver la Sección "Sobredosificación"). El tiempo de tromboplastina parcial activada (TTPa) y el HepTest también están prolongados, de manera dependiente de la dosis; sin embargo, no se recomiendan para evaluar el efecto farmacodinámico del rivaroxabán. La actividad anti-factor Xa también es afectada por el rivaroxabán; sin embargo, no se dispone de un patrón para la calibración. No es necesario vigilar los parámetros de la coagulación durante el tratamiento con rivaroxabán. El tiempo de tromboplastina parcial activada (TTPa) y el HepTest también están prolongados de manera dependiente de la dosis; sin embargo, no se los recomienda para evaluar el efecto farmacodinámico de rivaroxaban. No es necesario monitorizar los parámetros de coagulación durante el tratamiento clínico de rutina con Xarelto. Sin embargo, si se indica clínicamente, las concentraciones de rivaroxaban se pueden medir mediante pruebas del antifactor Xa cuantitativas calibradas (ver la Sección "Propiedades farmacocinéticas"). Eficacia clínica y seguridad: Prevención de la tromboembolia venosa en pacientes sometidos a una intervención de cirugía ortopédica mayor de las extremidades inferiores. El programa clínico del rivaroxabán se diseńo para demostrar la eficacia del rivaroxabán para la prevención de los incidentes de tromboembolia venosa, es decir, la trombosis venosa profunda, proximal y distal, y la embolia pulmonar en los pacientes sometidos a una intervención de cirugía ortopédica mayor de las extremidades inferiores. En estudios clínicos de fase III, controlados, aleatorizados y doble ciegos, el programa RECORD, se estudió a más de 9.500 pacientes (7.050 con cirugía de reemplazo total de la cadera y 2.531 con reemplazo total de la rodilla). El tratamiento con Xarelto, a una dosis de 10 mg 1 vez al día, iniciado no antes de 6 horas después de la intervención quirúrgica, se comparó con la enoxaparina, a una dosis de 40 mg 1 vez al día, iniciado 12 horas antes de la intervención. En los 3 estudios de fase III (véase la Tabla 2), el rivaroxabán redujo significativamente la tasa de incidentes de tromboembolia venosa (cualquier trombosis venosa profunda sintomática o detectada venográficamente, embolia pulmonar no mortal o muerte) e incidentes de tromboembolia venosa mayor (trombosis venosa profunda proximal, embolia pulmonar no mortal y muerte relacionada con incidentes de tromboembolia venosa), los criterios principales y secundarios mayores preexistentes de valoración de la eficacia. Además, en los 3 estudios, la tasa de incidentes de tromboembolia venosa sintomática (trombosis venosa profunda sintomática, embolia pulmonar no mortal, muerte relacionada con incidentes de tromboembolia venosa) fue más baja en los pacientes tratados conxarelto, en comparación con los pacientes tratados con enoxaparina. El principal criterio de valoración de la eficacia, la hemorragia mayor, mostró tasas comparables en los pacientes tratados con 10 mg de Xarelto, en comparación con 40 mg de enoxaparina.
El análisis de los resultados agrupados de los ensayos clínicos de fase III corroboró los datos obtenidos en los estudios individuales en cuanto a la reducción del total de los incidentes de tromboembolia venosa, incidentes de tromboembolia venosa mayor e incidentes de tromboembolia venosa sintomáticos con 10 mg de Xarelto, 1 vez al día, todos los días, en comparación con 40 mg de enoxaparina 1 vez al día, todos los días. Además del programa RECORD de fase III se realizó un estudio abierto de cohortes, de no intervención posterior a la autorización (XAMOS) en 17413 pacientes que sometidos a una cirugía ortopédica mayor de cadera o rodilla, para comparar rivaroxaban con otros tipos de tromboprofilaxis farmacológica estándar en un entorno de la vida real. Se produjo TEV sintomático en 57 (0.6%) pacientes en el grupo del rivaroxaban (n=8778) y en 88 (1.0%) pacientes en el grupo del estándar de atención (n=8635; IR 0.63; IC del 95% 0.43-0.91); población de seguridad. Se produjo sangrado mayor en 35 (0.4%) y 29 (0.3%) pacientes en los grupos de rivaroxaban y del estándar de tratamiento (IR 1.10; IC del 95% 0.67-1.80). Este estudio de no intervención confirmó los resultados de eficacia y seguridad que se obervaron en el programa RECORD. Un subanálisis post hoc del estudio abierto de cohortes, de no intervención posterior a la autorización (XAMOS) incluyó 790 pacientes que se sometían a una cirugía relacionada con una fractura de miembros inferiores que recibían rivaroxaban u otra tromboprofilaxis farmacológica estándar. Se produjo TEV sintomático en 2 (0.6%) pacientes en el grupo de rivaroxaban (n=350) y en 5 (1.1%) de los pacientes del grupo del estándar de tratamiento (n=440; IR 0.51; IC del 05% 0.10-2.61; población de seguridad). Se produjo sangrado mayor en 1 (0.3%) y 2 (0.5%) de los pacientes en los grupos del rivaroxaban y del estándar de atención (IR 0.97; IC del 95% 0.06-15.53). En este subanálisis, las incidencias de TEV y sangrado mayor en pacientes que se sometían a cirugía de los miembros inferiores relacionada con una fractura son comparables a las incidencias observadas en el programa RECORD en pacientes con reemplazo electivo de cadera y rodilla. Poblaciones especiales de pacientes:Diferencias étnicas: (Véase Propiedades farmacocinéticas). Personas de edad avanzada: (Véase Propiedades farmacocinéticas). Sexo del paciente: (Véase Propiedades farmacocinéticas).Categorías de pesos diferentes: (Véase Propiedades farmacocinéticas). Disfunción hepática: (Véase Propiedades farmacocinéticas). Disfunción renal: (Véase Propiedades farmacocinéticas). Efecto sobre el intervalo QTc: No se observó ningún efecto de prolongación del intervalo QTc en varones y mujeres sanos, mayores de 50 ańos. Propiedades farmacocinéticas: Absorción y biodisponibilidad: La biodisponibilidad absoluta del rivaroxabán es alta (80% - 100%), para la dosis en comprimidos de 2.5 mg y 10 mg independiente de las condiciones de ayuno/alimento. El rivaroxabán se absorbe rápidamente y alcanza concentraciones máximas (Cmax) de 2 a 4 horas después de tomar el comprimido. La toma con alimento no afecta el ABC ni la Cmax de rivaroxabán a la dosis de 10 mg. Los comprimidos de 2.5 y 10 mg de Xarelto puede tomarse con o sin alimento (véase Posología y metodo de administración). La variabilidad de las propiedades farmacocinéticas del rivaroxabán es moderada con una variabilidad interindividual (CV %) entre el 30% y el 40%. La absorción de rivaroxabán depende del sitio de liberación del medicamento en el tubo digestivo. Se reportó una disminución del 29% y del 56% en el ABC y Cmáx en comparación con el comprimido cuando se libera rivaroxabán granulado en el intestino delgado proximal. La exposición disminuye adicionalmente si se libera el medicamento en la porción distal del intestino delgado o en el colon ascendente. Evítese la administración de rivaroxabán distal al estómago, ya que puede disminuir la absorción y la exposición correspondiente al medicamento. La biodisponibilidad (ABC y Cmáx) de 20 mg de rivaroxabán administrados por vía oral como comprimido triturado y mezclado en puré de manzana o suspendido en agua y administrado por sonda nasogástrica y seguido de una comida líquida, fue comparable a la del comprimido entero. Dado el perfil farmacocinético predecible y proporcional a la dosis de rivaroxabán, los resultados de biodisponibilidad de este estudio probablemente sean aplicables a dosis más bajas de rivaroxabán. Distribución: En los seres humanos, la unión a las proteínas plasmáticas es alta, de aproximadamente el 92% al 95% y la albúmina sérica es el principal componente de unión. El volumen de distribución es moderado, el Vss es de aproximadamente 50 litros. Metabolismo y eliminación: De la dosis administrada de rivaroxabán, aproximadamente 2/3 se eliminan por degradación metabólica, siendo la mitad eliminado renalmente y la otra mitad se elimina por vía fecal. El otro 1/3 de la dosis administrada se elimina por excreción directa renal como principio activo inalterado en la orina, principalmente por secreción renal activa. El rivaroxabán es metabolizado mediante el CYP3A4, CYP2J2 y mecanismos independientes del CYP. La degradación oxidativa del grupo morfolinona y la hidrólisis de los ensalces amida son los sitios principales de biotransformación. En base a las investigaciones in vitro, el rivaroxabán es un sustrato de las proteínas transportadoras P-gp (glucoproteína P) y Bcrp (proteína de resistencia al cáncer de mama). El rivaroxabán inalterado es el compuesto más importante en el plasma humano, sin presencia de metabolitos circulantes principales activos. Con una depuración sistémica de alrededor de 10 l/h, el rivaroxabán puede clasificarse como fármaco de baja depuración. La eliminación de rivaroxabán del plasma se produjo con una vida media terminal de 5 a 9 horas en individuos jóvenes y con una vida media terminal de 11 a 13 horas en personas de edad avanzada. Pacientes geriátricos: Los pacientes de edad avanzada presentaron concentraciones plasmáticas más altas que los pacientes más jóvenes, y los valores promedios del ABC fueron aproximadamente 1.5 veces más altos, principalmente debido a la disminución (aparente) de la depuración total y renal (véase Posología y Método de administración). Sexo: No hubo ninguna diferencia clínicamente relevante en las propiedades farmacocinéticas entre los pacientes varones y las mujeres (véase Posología y Método de administración). Categorías de pesos diferentes: Los extremos en el peso corporal (< 50 kg en comparación con > 120 kg) sólo tuvieron un efecto pequeńo en las concentraciones plasmáticas de rivaroxabán (menos del 25%) (véase Posología y Método de administración). Nińos y adolescentes: No se han establecido la seguridad y la eficacia en nińos y adolescentes menores de 18 ańos (véase Posología y Método de administración). Diferencias interétnicas: No se observaron diferencias interétnicas clínicamente relevantes entre los pacientes de raza caucásica, afro-americana, hispana, japonesa o china, en cuanto a las propiedades farmacocinéticas o farmacodinámicas (véase Posología y Método de administración). Disfunción hepática: Se ha estudiado el efecto de la disfunción hepática en la farmacocinética de rivaroxabán en sujetos categorizados según la clasificación de Child Pugh, un procedimiento estándar en el desarrollo clínico. El objetivo original de la clasificación de Child Pugh es evaluar el pronóstico de la hepatopatía crónica, principalmente la cirrosis. En los pacientes a los que se pretende administrar anticoagulantes, el aspecto crítico de la insuficiencia hepática es la síntesis reducida de factores de la coagulación normal en el hígado. Debido a que este aspecto es reflejado sólo por 1 de las 5 determinaciones clínicas/bioquímicas que constituyen el sistema de clasificación de Child Pugh, el riesgo de hemorragia en pacientes no puede correlacionarse claramente con este esquema de clasificación. Por tanto, la decisión de tratar a pacientes con un anticoagulante debe tomarse independientemente de la clasificación de Child Pugh. Xarelto 10 mg está contraindicado en pacientes con enfermedad hepática asociada con coagulopatía conducente a un riesgo clínicamente relevante de hemorragia. Los pacientes con cirrosis y con disfunción hepática leve (clasificados como Child Pugh A) sólo presentaron cambios menores en las propiedades farmacocinéticas del rivaroxabán (aumento de 1.2 veces el ABC del rivaroxabán en promedio), lo que es casi comparable a su grupo de control sano apareado. No se observó ninguna diferencia de importancia en las propiedades farmacodinámicas entre estos grupos. En los pacientes con cirrosis y con disfunción hepática moderada (clasificados como Child Pugh B), el ABC media del rivaroxabán estaba aumentado significativamente en 2.3 veces, en comparación con los voluntarios sanos, debido a una depuración significativamente alterada del fármaco, lo que indica una enfermedad hepática significativa. El ABC no fijado aumentó 2.6 veces. No existen datos en pacientes con insuficiencia hepática severa. La inhibición de la actividad del factor Xa aumentó en un factor de 2.6 en comparación con voluntarios sanos; de manera similar, la prolongación del TP también aumentó, en un factor de 2.1. El TP, una prueba de coagulación total, evalúa la vía extrínseca, que comprende los factores de la coagulación VII, X, V, II y I, que se sintetizan en el hígado. Los pacientes con disfunción hepática moderada eran más sensibles a rivaroxabán, dando lugar a una relación FC/FD más pronunciada entre la concentración y el TP. No se dispone de datos en cuanto a los pacientes Child Pugh C (véase Posología y Método de administración y Contraindicaciones). Disfunción renal: Hubo un aumento de la exposición del rivaroxabán, que se correlacionó de manera inversa con la disminución de la función renal, evaluada mediante las determinaciones de la depuración de creatinina. En las personas con disfunción renal leve (CrC• de 80 a 50 ml/min), moderada (CrC < 50 a 30 ml/min) o grave (CrC < 30-15 ml/min), las concentraciones plasmáticas de rivaroxabán (ABC) estuvieron aumentadas 1.4, 1.5, y 1.6 veces, respectivamente, en comparación con voluntarios sanos (véase Posología y Método de administración y Advertencias y precauciones especiales de empleo). Los aumentos correspondientes de los efectos farmacodinámicos fueron más pronunciados (véase Posología y Método de administración y Advertencias y precauciones especiales de empleo). En las personas con disfunción renal leve, moderada o grave, la inhibición total de la actividad del factor Xa aumentó en un factor de 1.5; en la disfunción renal moderada, en un factor de 1.9, y en la disfunción renal grave, del 2.0, en comparación con voluntarios sanos; de manera similar, la prolongación del TP aumentó, respectivamente, en factores de 1.3, 2.2 y 2.4. No hay datos de pacientes con CrC < 15 ml/min. Debido a la enfermedad de fondo, los pacientes con disfunción renal grave tienen un mayor riesgo de hemorragia y de trombosis. Datos preclínicos sobre seguridad: Salvo los efectos relacionados con un modo de acción farmacológico exagerado (hemorragias), los datos preclínicos no revelan ningún peligro especial para los seres humanos, según los estudios sobre farmacología de la seguridad, toxicidad con dosis repetidas y genotoxicidad. Se examinó la seguridad no clínica del rivaroxaban en estudios convencionales y adecuados de farmacología de la seguridad, toxicidad con dosis única y repetidas, genotoxicidad, fototoxicidad y toxicidad para la reproducción. No se observó ninguna toxicidad específica de órganos del rivaroxaban hasta la dosis más alta estudiada. Farmacología de seguridad: Las funciones cardiovasculares, respiratorias y del SNC no se afectaron. No se observó ningún potencial proarrítmico. No se observó ningún efecto clínicamente relevante sobre la motilidad gastrointestinal, la función hepática, la función renal ni la glucemia. Toxicidad con dosis agudas y repetidas: El rivaroxabán mostró una toxicidad aguda baja en ratas y ratones. El rivaroxabán se examinó en estudios con dosis repetidas, hasta 6 meses, en ratas, y hasta 12 meses, en perros. A partir del modo de acción farmacológico, no se pudo establecer un NOEL, debido a los efectos sobre el tiempo de coagulación. Todos los resultados adversos, salvo una ligera reducción del aumento del peso en ratas y perros, pudieron estar relacionados con un modo de acción farmacológico exagerado del compuesto. En perros, a exposiciones muy altas, se observaron hemorragias espontáneas graves. Los NOAEL después de la exposición crónica son 12.5 mg/kg en ratas, y 5 mg/kg en perros. Carcinogénesis: El rivaroxabán se investigó hasta 60 mg/kg/día, alcanzando niveles de exposición similares a los de seres humanos (ratones) o hasta 3.6 veces más (ratas) que en seres humanos. El rivaroxabán no demostró potencial carcinogénico en ratas y ratones. Toxicidad reproductiva: Se examinó el rivaroxabán en estudios de toxicidad del desarrollo, a niveles de exposición de hasta 14 veces (en la rata) y de hasta 33 veces (en el conejo) superiores a la exposición terapéutica en los seres humanos. El perfil toxicológico se caracteriza principalmente por toxicidad materna debido a efectos farmacodinámicos exagerados. Hasta la dosis más alta investigada, no se identificó ningún potencial teratogénico primario (ver sección Embarazo y lactancia). La radiactividad relacionada con el rivaroxabán marcado con [14C] penetró la barrera placentaria en ratas. En ninguno de los órganos y tejidos fetales, la exposición, en cuanto a las concentraciones máximas o ABC, sobrepasó la exposición en la sangre materna. La exposición promedio en los fetos, basada en el ABC (0-24), alcanzó aproximadamente el 20% de la exposición en la sangre materna. Las glándulas mamarias tuvieron un ABC aproximadamente equivalente al de la sangre, lo que indica la secreción de radiactividad hacia la leche (véase Embarazo y lactancia). El rivaroxabán no mostró ningún efecto sobre la fertilidad masculina o femenina, hasta 200 mg/kg (véase Embarazo y lactancia). Lactancia: El rivaroxabán marcado con [14C] se administró por vía oral a ratas de Wistar en lactancia (8 a 10 días después del parto) como dosis única, por vía oral, de 3 mg/kg de peso corporal. La radiactividad relacionada con el rivaroxabán marcado con [14C] se secretó por la leche de ratas en lactación, sólo en menor grado, en relación con la dosis administrada: la cantidad estimada de radiactividad excretada por la leche fue del 2.12% de la dosis materna, en las 32 horas siguientes a la administración (véase Embarazo y lactancia). Genotoxicidad: No se observó genotoxicidad en una prueba de mutación génica en bacteria (prueba de Ames), en prueba in vitro de aberraciones cromosómicas, ni en la prueba de micronúcleos in vivo.
Posología:Método de administración: Vía oral. Dosis habitual recomendada: La dosis recomendada para la prevención de la tromboembolia venosa en las intervenciones quirúrgicas ortopédicas mayores es de 1 comprimido de 10 mg 1 vez al día. Duración del tratamiento: La duración del tratamiento depende del tipo de intervención quirúrgica ortopédica mayor. Después de la cirugía mayor de cadera, los pacientes deben recibir tratamiento durante 5 semanas. Después de la cirugía mayor de rodilla, los pacientes deben recibir tratamiento durante 2 semanas. Forma y frecuencia de administración: 1 comprimido de 10 mg de Xarelto se debe tomar 1 vez al día. Xarelto puede tomarse con o sin alimentos. La dosis inicial deberá administrarse de 6 a 10 horas después del final de la intervención quirúrgica, siempre que se haya restablecido la hemostasia. Si la dosis se omite, el paciente debe tomar Xarelto inmediatamente y continuar al día siguiente con la toma 1 vez al día, como antes. En pacientes que no puedan tragar comprimidos enteros, se puede triturar el comprimido de Xarelto y mezclar con agua o algún alimento blando como puré de manzana inmediatamente antes de su uso y administración por vía oral. El comprimido triturado de Xarelto se puede administrar por sonda nasogástrica. La colocación correcta de la sonda nasogástrica en el estómago se debe confirmar antes de administrar Xarelto. El comprimido triturado se debe administrar en un volumen pequeńo de agua por la sonda nasogástrica, irrigándola posteriormente con agua para su vaciado en el estómago. (ver sección Propiedades farmacocinéticas). Dosis olvidadas: Si una dosis se omite, el paciente debe tomar la dosis de 10 mg de Xarelto inmediatamente y continuar al día siguiente con la toma 1 vez al día, como antes. Información adicional sobre poblaciones especiales: Nińos y adolescentes: No se dispone de datos clínicos en cuanto a los nińos. Pacientes geriátricos: No se requiere ningún ajuste de la dosis para estas poblaciones de pacientes (véase Propiedades farmacocinéticas). Sexo: No se requiere ningún ajuste de la dosis para estas poblaciones de pacientes (véase Propiedades farmacocinéticas). Peso corporal: No se requiere ningún ajuste de la dosis para estas poblaciones de pacientes (véase Propiedades farmacocinéticas). Pacientes con disfunción hepática: Xarelto está contraindicado en los pacientes con enfermedad hepática asociada a una coagulopatía que lleva a un riesgo clínicamente relevante de hemorragia. No es necesario ningún ajuste a la dosis en los pacientes con otras enfermedades hepáticas (véase Propiedades farmacocinéticas). Los datos clínicos limitados en los pacientes con disfunción hepática moderada (Child Pugh B) indican un aumento significativo de la actividad farmacológica. No se dispone de datos clínicos en los pacientes con disfunción hepática grave (Child Pugh C) (véase Contraindicaciones y Propiedades farmacocinéticas). Pacientes con disfunción renal: No se requiere ningún ajuste de la dosis si Xarelto se administra en los pacientes con disfunción renal leve (CrC:• 80 a 50 ml/min (o moderada (CrC: <50 a 30 ml/min) (véase Propiedades farmacocinéticas). Los limitados datos clínicos en los pacientes con disfunción renal grave (CrC): <30-15 ml/min) indican que las concentraciones plasmáticas del rivaroxabán están aumentadas significativamente en esta población de pacientes. Por lo tanto, Xarelto debe emplearse con precaución en estos pacientes. El uso de rivaroxabán no se recomienda en pacientes con CrC <15 ml/min (véase Advertencias y precauciones especiales de empleo y Propiedades farmacocinéticas). Diferencias étnicas: No se requiere ningún ajuste de la dosis según las diferencias étnicas (véase Propiedades farmacocinéticas).
Modo de Empleo:Instrucciones de uso/manipulación: Ninguna.
Efectos Colaterales: Se ha evaluado la seguridad de Xarelto en 4 estudios de fase III, con 6097 pacientes expuestos a 10 mg de Xarelto sometidos a cirugía ortopédica mayor de las extremidades inferiores (reemplazo total de la cadera o reemplazo total de la rodilla), en 3997 pacientes médicamente enfermos y hospitalizados tratados durante un período de hasta 39 días, y en 3 ensayos de fase III de tratamiento del TEV con 4556 pacientes expuestos a 15 mg de Xarelto 2 veces al día durante 3 semanas seguidos de 20 mg 1 vez al día o expuestos a 20 mg 1 vez al día, tratados durante un período de hasta 21 meses. En total, se reportó que cerca del 67% de los pacientes expuestos a por lo menos una dosis de rivaroxabán presentaron eventos adversos emergentes del tratamiento. Alrededor del 22% de los pacientes experimentaron eventos adversos que se consideran relacionados con el tratamiento, según la evaluación de los investigadores. En los pacientes tratados con 10 mg de Xarelto sometidos a la cirugía de remplazo de cadera o rodilla y en los pacientes enfermos hospitalizados, los eventos hemorrágicos ocurrieron en aproximadamente el 6.8% y el 8.8% de los pacientes respectivamente; y la anemia ocurrió en alrededor del 5.9% y 2.1% de los pacientes, respectivamente. Debido al modo de acción farmacológico, Xarelto puede asociarse a un aumento del riesgo de hemorragia oculta o manifiesta de cualquier tejido y órgano, que puede producir anemia post-hemorrágica. Los signos, síntomas y gravedad (incluso posible desenlace mortal) variarán según la localización y el grado o la magnitud de la hemorragia y/o anemia. Las complicaciones hemorrágicas pueden presentarse como debilidad, palidez, mareos, cefalea o hinchazón inexplicada, disnea y schock inexplicado. En algunos casos, pueden producirse síntomas de isquemia cardíaca, como dolor torácico o angina de pecho, como consecuencia de la anemia. Por lo tanto, al evaluar el estado de cualquier paciente anticoagulado deberá plantearse la posibilidad de una hemorragia. Las frecuencias de las RAM notificadas con Xarelto se resumen en la tabla siguiente. Dentro de cada grupo de frecuencia, las reacciones adversas se presentan en orden decreciente de gravedad. Las frecuencias se definen como: frecuentes (≥ 1/100 a < 1/10), poco frecuentes (≥ 1/1.000 a < 1/100), raras (≥ 1/10.000 a < 1/1.000). Las RAM identificadas sólo durante la vigilancia postcomercialización y cuya frecuencia no pudo estimarse, se clasifican como "no conocida". Trastornos de la sangre y del sistema linfático: Frecuentes: Anemia (incl. parámetros de laboratorio respectivos). Poco frecuentes: Trombocitosis (incl. aumento del recuento de plaquetas). Trastornos cardíacos: Poco frecuentes: Taquicardia. Trastornos oculares: Frecuentes: Hemorragia ocular (incl. hemorragia conjuntival): Trastornos gastrointestinales: Frecuentes: Náuseas, hemorragia gingival, hemorragia del tracto gastrointestinal (incl. hemorragia rectal), dolores gastrointestinales y abdominales, disepsia, estreńimiento, diarrea, vómitos. Poco frecuentes: Sequedad de boca. Trastornos generales y alteraciones en el lugar de administración: Frecuentes: Fiebre. Edema periférico. Poco frecuentes: Sensación de malestar. Raros: Edema localizado. Trastornos hepatobiliares: Raros: Función hepática anómala. Trastornos del sistema inmunológico: Raros: Dermatitis alérgica. Lesiones traumáticas, intoxicaciones y complicaciones tras procedimientos terapéuticos: Frecuentes: Hemorragia post procedimiento (incl. anemia postoperatoria y hemorragia de la herida). Poco frecuentes: Secreción de la herida. Exploraciones complementarias: Frecuentes: Aumento de GGT. Aumento de las transaminasas. Poco frecuentes: Aumento de la lipasa. Aumento de la amilasa. Aumento de la bilirrubina sanguínea. Aumento de la LDH. Aumento de la fosfatasa alcalina. Raros: Aumento de la bilirrubina conjugada (con o sin aumento concomitante de la ALT). Trastornos musculoesqueléticos, del tejido conjuntivo y de los huesos: Poco frecuentes: Dolor en las extremidades. Trastornos del sistema nervioso:Frecuentes: Mareos. Cefalea. Poco frecuentes: Síncope (incl. pérdida de la conciencia). Trastornos renales y urinarios: Poco frecuentes: Disfunción renal (incl. aumento de la creatinina en la sangre, aumento de la urea en la sangre). Trastornos de la piel y del tejido subcutáneo: Poco frecuentes: Prurito (incl. casos poco frecuentes de prurito generalizado). Contusión. Raros: Urticaria (incl. Casos raros de prurito generalizado. Trastornos vasculares: Poco frecuentes: Hipotensión (incl. Disminución de la presión arterial, hipotensión intrahospitalaria). Hematoma (incl. Casos raros de hemorragia muscular). Hemorragia del tubo digestivo (incl. hemorragia gingival, hemorragia rectal, hematemesis). Hemorragia del tracto urogenital. Epistaxis. < La representación terminológica de las RAM se basa en la versión 12 del MedDRA. En otros estudios clínicos con el rivaroxabán, se notificaron casos únicos de hemorragia suprarrenal y hemorragia conjuntival, y hemorragia mortal por úlcera gastrointestinal; la ictericia y la hipersensibilidad fueron raras y la hemoptisis fue poco frecuente. Se ha descrito la hemorragia intracraneal (especialmente en los pacientes con hipertensión arterial, con antihemostáticos concomitantes o con ambas condiciones) que, en algunos casos aislados, puede poner en peligro la vida del paciente. En otros estudios clínicos, se ha notificado la formación de pseudoaneurismas vasculares tras la realización de intervenciones percutáneas. Además, en otros estudios clínicos y de vigilancia post-comercialización, se han notificado complicaciones conocidas secundarias a las hemorragias, como el síndrome compartimental. También se han notificado casos de insuficiencia renal aguda/insuficiencia renal secundarios a un acontecimiento hemorrágico de magnitud suficiente para causar hipoperfusión. Observaciones posteriores a la comercialización: Se han reportado casos de angioedema y edema alérgico posterior a la comercialización, en asociación temporal con el uso de Xarelto. No es posible calcular la frecuencia de estos eventos adversos informados en base a la experiencia posterior a la comercialización. En los ensayos combinados de fase III, estos eventos fueron poco frecuentes (≥1/1000 a < 1/100).
Contraindicaciones: Xarelto está contraindicado en los pacientes con hipersensibilidad al rivaroxabán o a cualquier excipiente del comprimido (véase Datos farmacéuticos). Xarelto está contraindicado en los pacientes con hemorragia activa, clínicamente significativa (por ej.: hemorragia intracraneal, hemorragia gastrointestinal). Xarelto está contraindicado en los pacientes con enfermedad hepática que se asocie a coagulopatía que lleve a un riesgo clínicamente relevante de hemorragia. No se dispone de datos en seres humanos en cuanto al uso de Xarelto en mujeres embarazadas. Los datos en animales demuestran que el rivaroxabán atraviesa la barrera placentaria. Por lo tanto, el uso de Xarelto está contraindicado durante el embarazo (véase Embarazo y lactancia y Datos preclínicos sobre seguridad). No se dispone de datos en seres humanos en cuanto al uso de Xarelto en las mujeres en lactación. Los datos en animales indican que el rivaroxabán se secreta por la leche materna. Por lo tanto, Xarelto está contraindicado durante la lactancia (véase Embarazo y lactancia y Datos preclínicos sobre seguridad).
Advertencias:Advertencias y precauciones especiales de empleo: Medicación concomitante: Xarelto no está recomendado en pacientes que reciben tratamiento sistémico concomitante con antimicóticos azólicos (por ej. ketoconazol o inhibidores de la proteasa del HIV (por ej. ritonavir). Estos fármacos son potentes inhibidores de CYP 3A4 y P-gp. Por tanto, estos fármacos pueden aumentar las concentraciones plasmáticas de rivaroxabán hasta un grado clínicamente relevante que puede ocasionar un riesgo aumentado de hemorragia (véase Interacciones con otros medicamentos y otras formas de interacción). Insuficiencia renal: Xarelto se ha de usar con precaución en pacientes con insuficiencia renal moderada (depuración de creatinina 30-49 ml/min) que reciben comedicación que ocasiona concentraciones plasmáticas aumentadas de rivaroxabán (véase Interacciones con otros medicamentos y otras formas de interacción). En pacientes con insuficiencia renal grave (CrC: <30 ml/min), las concentraciones plasmáticas de rivaroxabán pueden aumentar significativamente y ocasionar un riesgo aumentado de hemorragia. Debido a la enfermedad subyacente, estos pacientes tienen un riesgo aumentado de hemorragia y trombosis. Debido a datos clínicos limitados, rivaroxabán debe usarse con precaución en pacientes con CrC <30-15 ml/min. No hay datos clínicos disponibles de pacientes con insuficiencia renal grave (CrC <15 ml/min. Por tanto, no se recomienda el uso de Xarelto en estos pacientes (véase Posología, Propiedades farmacocinéticas y Propiedades farmacodinámicas). Los pacientes con insuficiencia renal grave o riesgo hemorrágico aumentado y los pacientes que reciben tratamiento sistémico concomitante con antimicóticos azólicos o inhibidores de la proteasa de HIV se han de monitorizar cuidadosamente en cuanto a signos de complicaciones hemorrágicas después de la iniciación del tratamiento (ver sección Interacción con otros medicamentos y otras formas de interacción). Esto puede realizarse por exámenes físicos regulares de los pacientes, observación estrecha del drenaje de la herida quirúrgica y determinaciones periódicas de hemoglobina. Cirugía de fractura de cadera: Xarelto no se ha estudiado en ensayos clínicos con pacientes sometidos a cirugía de fractura de cadera. Riesgo de hemorragia: Xarelto, al igual que otros antitrombóticos, deberá emplearse con precaución en los pacientes con un aumento del riesgo de hemorragia, por ejemplo: Trastornos hemorrágicos congénitos o adquiridos. Hipertensión arterial grave y no controlada. Enfermedad gastrointestinal ulcerosa activa. Ulceraciones gastrointestinales recientes. Retinopatía vascular. Hemorragia intracraneal o intracerebral reciente. Anormalidades vasculares intracerebrales o intrarraquídeas. Poco después de una intervención quirúrgica cerebral, vertebral u oftalmológica. Debe tenerse precaución si los pacientes reciben tratamiento concomitante con fármacos que afectan a la hemostasia, como los antiinflamatorios no esteroides (AINEs), los inhibidores de la agregación plaquetaria u otros antitrombóticos (véase Interacciones con otros medicamentos y otras formas de interacción). Cualquier descenso inexplicado de la hemoglobina o de la presión arterial deberá llevar a una búsqueda de la localización de la hemorragia. Anestesia neuroaxial (epidural/medular): Cuando se aplica anestesia neuroaxial (epidural/medular) o se realiza una punción lumbar en los pacientes tratados con antitrombóticos para la prevención de complicaciones tromboembólicas, tienen riesgo de presentar un hematoma epidural o medular, que puede causar parálisis a largo plazo. El riesgo de estos incidentes aumenta más incluso por el uso de catéteres epidurales permanentes o por el uso concomitante de fármacos que afectan a la hemostasia. El riesgo también puede aumentar con la punción epidural o lumbar traumática o repetida. Se debe vigilar con frecuencia en los pacientes la presencia de signos y síntomas de trastorno neurológico (por ejemplo, adormecimiento o debilidad de las extremidades inferiores o disfunción intestinal o vesical). Si se observan deficiencias neurológicas, son necesarios el diagnóstico y el tratamiento urgentes. El médico deberá tener en cuenta el posible beneficio frente al riesgo de intervención neuroaxial en los pacientes con tratamiento anticoagulante o que van a recibir anticoagulantes para tromboprofilaxis. Un catéter epidural no deberá retirarse antes de 18 horas después de la última administración de Xarelto. Xarelto deberá administrarse, como mínimo, 6 horas después de la retirada del catéter. Si se produce una punción traumática, la administración del rivaroxabán deberá retrasarse 24 horas. Mujeres en edad fértil: Xarelto deberá utilizarse en las mujeres en edad fértil sólo con medidas anticonceptivas eficaces. Prolongación del intervalo QTc: No se ha observado un efecto de prolongación del intervalo QTc con Xarelto. Información sobre los excipientes: Dado que este medicamento contiene lactosa, los pacientes con problemas hereditarios y poco frecuentes de intolerancia a la lactosa o a la galactosa (por ejemplo, deficiencia de lactasa de los lapones o mal absorción de glucosa -galactosa) no deberían tomar Xarelto.
Precauciones:Embarazo y lactancia: Embarazo: No se dispone de datos en seres humanos en cuanto al uso de Xarelto en mujeres embarazadas. En ratas y conejos, el rivaroxabán demostró una toxicidad materna pronunciada, con alteraciones placentarias relacionadas con su modo de acción farmacológico (por ejemplo, complicaciones hemorrágicas). No se ha identificado ningún potencial teratógeno primario. Los datos en animales demostraron una toxicidad materna marcada de rivaroxabán con respecto a su modo de acción farmacológico (por ejemplo, complicaciones hemorrágicas) que ocasionaron toxicidad en la reproducción (véase Datos preclínicos sobre seguridad). Debido al riesgo intrínseco de hemorragia y a la evidencia de que rivaroxabán atraviesa la placenta, Xarelto está contraindicado en el embarazo (véase Contraindicaciones y Datos preclínicos sobre seguridad). Mujeres en edad fértil/anticoncepción: Xarelto deberá utilizarse en las mujeres en edad fértil sólo con medidas anticonceptivas eficaces. Lactancia: No se dispone de datos en seres humanos en cuanto al uso de Xarelto en las mujeres en lactación. En las ratas, el rivaroxabán se secreta por la leche materna. Por lo tanto, Xarelto sólo puede administrarse después de interrumpir la lactancia materna (véase Contraindicaciones y Datos preclínicos sobre seguridad). Efectos sobre la capacidad de conducir o utilizar máquinas: No se ha comunicado ningún efecto del rivaroxabán sobre la capacidad de conducir o utilizar máquinas.
Interacciones Medicamentosas:Interacción con otros medicamentos y otras formas de interacción: Interacciones farmacocinéticas: El rivaroxabán se depura principalmente por medio del metabolismo hepático, mediado por el citocromo P450 (CYP 3A4, CYP 2J2), y la excreción del fármaco no modificado, en que intervienen los sistemas transportadores de P-glucoproteína (P-gp)/y de la proteína de resistencia al cáncer de mama (Bcrp). Inhibición del CYP: El rivaroxabán no inhibe el CYP 3A4 ni ninguna otra isoforma mayor del CYP. Inducción del CYP: El rivaroxabán no induce el CYP 3A4 ni ninguna otra isoforma mayor del CYP. Efectos sobre rivaroxabán: El uso concomitante de Xarelto con inhibidores potentes del CYP 3A4 y de la P-gp puede llevar a una disminución de la depuración hepática y renal y, por lo tanto, puede aumentar significativamente la exposición sistémica. La administración concomitante de Xarelto con el ketoconazol, un antimicótico azólico (400 mg 1 vez al día), un inhibidor potente del CYP 3A4 y de la P-gp, produjo un aumento de 2.6 veces del ABC media del rivaroxabán, en estado de equilibrio, y un aumento 1.7 veces de la Cmax media del rivaroxabán, con aumentos significativos de sus efectos farmacodinámicos. La administración concomitante del rivaroxabán con ritonavir, un inhibidor de la proteasa del VIH (600 mg cada 12 horas), un inhibidor potente del CYP 3A4 y de la P-gp, produjo un aumento de 2.5 veces el ABC media del rivaroxabán y un aumento de 1.6 veces de la Cmax media del rivaroxabán, con aumentos significativos de sus efectos farmacodinámicos. Por lo tanto, el rivaroxabán no está recomendado en los pacientes que reciben tratamiento concomitante, por vía sistémica con antimicóticos azólicos o inhibidores de la proteasa del VIH (véase Advertencias y precauciones especiales de empleo). Otros principios activos que inhiben potentemente sólo una de las vías de eliminación de rivaroxabán, el CYP 3A4 o la P-gp, son de esperar que aumenten las concentraciones plasmáticas de rivaroxabán en menor extensión. La claritromicina (500 mg 2 veces al día), considerada como inhibidora potente del CYP 3A4 e inhibidora moderada de la P-gp, ocasionó un aumento 1.5 veces del ABC media del rivaroxabán y un aumento 1.4 veces de la Cmax. Este aumento, próximo a la magnitud de la variabilidad normal de ABC y Cmax se considera no relevante clínicamente. La eritromicina (500 mg cada 8 horas), que inhibe moderadamente el CYP 3A4 y la P-gp, produjo un aumento de 1.3 veces de la ABC y la Cmax medias del rivaroxabán. Este aumento está dentro de la magnitud de la variabilidad normal del ABC y de la Cmax y se considera clínicamente no relevante. El fluconazol (400 mg 1 vez al día), considerado como inhibidor moderado del CYP 3A4, ocasionó un aumento 1.4 veces del ABC media de rivaroxabán y un aumento 1.3 veces de la Cmáx media. Este aumento está dentro de la magnitud de la variabilidad normal del ABC y de la Cmáx y se considera clínicamente no relevante (ver sección Advertencias y precauciones especiales de empleo). La administración concomitante del rivaroxabán con rifampicina, un potente inductor del CYP 3A4 y de la P-gp, produjo una disminución del 50% del ABC media del rivaroxabán, con disminuciones paralelas en sus efectos farmacodinámicos. El uso concomitante del rivaroxabán con otros inductores potentes del CYP 3A4 (por ejemplo, fenitoína, carbamazepina, fenobartirona, o hipérico) también puede causar una disminución de la concentración plasmática del rivaroxabán. La disminución de las concentraciones plasmáticas de rivaroxabán se considera clínicamente no importante. Interacciones farmacodinámicas: Después de la administración combinada de enoxaparina (dosis única de 40 mg) con el rivaroxabán (dosis única de 10 mg), se observó un efecto aditivo sobre la actividad anti-factor Xa, sin efectos adicionales en las pruebas de coagulación (TP, TTPa). La enoxaparina no afectó a las propiedades farmacocinéticas del rivaroxabán (véase Advertencias y precauciones especiales de empleo). El clopidogrel (dosis de carga de 300 mg y después, dosis de mantenimiento de 75 mg) no mostró ninguna interacción farmacocinética, pero se observó un aumento pertinente en los tiempos de sangrado en un subgrupo de pacientes que no se correlacionó con la agregación plaquetaria, ni las concentraciones de P-selectina ni de los receptores Gpllb/llla (véase Advertencias y precauciones especiales de empleo). No se ha observado ninguna prolongación clínicamente relevante del tiempo de sangrado después de la administración concomitante de rivaroxabán y 500 mg de naproxeno. No obstante, puede haber personas con una respuesta farmacodinámica más pronunciada (véase Advertencias y precauciones especiales de empleo). Alimentos y productos lácteos: 10 mg de Xarelto pueden tomarse con o sin alimentos (véase Propiedades farmacocinéticas). Interacciones que se han probado en la práctica: No hubo interacciones farmacocinéticas mutuas entre el rivaroxabán y el midazolam (sustrato del CYP 3A4), la digoxina (sustrato de la glucoproteína P) ni la atorvastatina (sustrato del CYP 3A4 y la P-gp). La administración concomitante de ranitidina, un antagonista de los receptores H2, el antiácido hidróxido de aluminio / hidróxido de magnesio, naproxeno, clopidogrel o enoxaparina no afectó a la biodisponibilidad ni a las propiedades farmacocinéticas del rivaroxabán. No se observó ninguna interacción farmacocinética ni farmacodinámica clínicamente significativa cuando se administró Xarelto concomitantemente con 500 mg de ácido acetilsalicílico. Interacciones con parámetros de laboratorio: Las pruebas de los parámetros de la coagulación (TP, TTPa, Hep Test) se afectan de la manera esperada por el modo de acción de Xarelto.
Sobredosificación: La sobredosis después de la administración de rivaroxabán puede causar complicaciones hemorrágicas debido a sus propiedades farmacodinámicas. No hay disponible ningún antídoto específico que antagonice el efecto farmacéutico de rivaroxabán. Puede plantearse el uso de carbono activado para reducir la absorción en caso de sobredosis por Xarelto. La administración de carbono activado hasta ocho horas después de las sobredosis puede reducir la absorción del rivaroxaban. Debido a la elevada fijación a las proteínas plasmáticas, no se espera que el rivaroxabán sea dializable. En caso de producirse hemorragia, su tratamiento consistirá en las siguientes medidas: Retraso de la siguiente administración de Xarelto o interrupción del tratamiento si se considera conveniente. El rivaroxabán tiene una vida media de aproximadamente 5 a 13 horas (véase "Propiedades farmacocinéticas"). Deberá plantearse el tratamiento sintomático adecuado, por ejemplo, compresión mecánica (por ejemplo, para la epistaxis grave), intervenciones quirúrgicas reemplazo hídrico y apoyo hemodinámico y transfusión de derivados o componentes hemáticos. Si la epistaxis no puede controlarse por las medidas anteriores, puede considerarse la administración de uno de los siguientes procoagulantes: Concentrado de complejo de protrombina activada (APCC). Concentrado de complejo de protrombina (PCC). Factor VIIa recombinante (r-FVIIa). Sin embargo, actualmente hay una experiencia con el uso de estos productos en las personas que reciben Xarelto. No se espera que el sulfato de protamina y la vitamina K afecten a la actividad anticoagulante de Xarelto. No hay una justificación científica sobre la ventaja o la experiencia con hemostáticos sistémicos (por ejemplo, desmopresina, aprotinina, ácido tranexámico, ácido aminocaproico) en las personas que reciben Xarelto.
Conservación: En cuanto a los comprimidos, no se aplican restricciones en cuanto a la conservación (temperatura, humedad, luz).
 Laboratorio
Laboratorio