Antiinfecciosos de Uso Sistémico: Antibióticos: Tetraciclinas
Información farmacológica
Composición: Formulación de Tigeciclina Lactosa: Tigeciclina para inyección se suministra en dosis única, en viales de vidrio tipo 1 de 5 ml con 50 mg de Polvo Liofilizado para Infusión. Cada vial contiene: 100 mg de Lactosa Monohidrato. El pH se ajusta con Ácido Clorhídrico y, de ser necesario, con Hidróxido de Sodio.
Indicaciones:Tygacil® está indicado para el tratamiento de infecciones causadas por cepas sensibles de los microorganismos citados en las condiciones listadas más abajo, para pacientes de 18 ańos y mayores: Infecciones complicadas de piel y estructuras de la piel causadas por Escherichia coli, Enterococcus faecalis (sólo aislados sensibles a la vancomicina), Staphylococcus aureus (aislados sensibles y resistentes a la meticilina), Streptococcus agalactiae, grupo Streptococcus anginosus (incluye S. anginosus, S. intermedius y S. constellatus), Streptococcus pyogenes y Bacteroides fragilis. Infecciones intraabdominales complicadas causadas por Citrobacter freundii, Enterobacter cloacae, Escherichia coli, Klebsiella oxytoca, Klebsiella pneumoniae, Enterococcus faecalis (sólo aislados sensibles a la vancomicina), Staphylococcus aureus (sólo aislados sensibles a la meticilina), grupo Streptococcus anginosus (incluye S. anginosus, S. intermedius y S. constellatus), Bacteroides fragilis, Bacteroides thetaiotaomicron, Bacteroides uniformis, Bacteroides vulgatus, Clostridium perfringens y Peptostreptococcus micros. Deberán obtenerse muestras adecuadas para análisis bacteriológicos para efectos de aislar e identificar los microorganismos patógenos y determinar su sensibilidad a tigeciclina. Tygacil® puede administrarse como tratamiento empírico inicial antes de conocer los resultados de estos análisis. Para reducir el desarrollo de bacterias resistentes al medicamento y mantener la eficacia de éste y otros antibióticos, Tygacil® deberá emplearse únicamente para tratar infecciones comprobadas o con alta probabilidad de ser causadas por bacterias sensibles. Una vez obtenidos el cultivo y la información de sensibilidad, los mismos deberán ser considerados para determinar o modificar el tratamiento antibacteriano. En ausencia de dichos datos, la epidemiología bacteriana y los patrones de sensibilidad locales contribuirán para la selección empírica del tratamiento.
Posología: El régimen posológico recomendado de tigeciclina es 1 dosis inicial de 100 mg, seguido de 50 mg cada 12 horas. Las infusiones intravenosas (IV) de tigeciclina deberán administrarse durante aproximadamente 30 a 60 minutos cada 12 horas. La duración recomendada del tratamiento con tigeciclina para infecciones complicadas de piel y de las estructuras cutáneas o para infecciones complicadas intraabdominales es de 5 a 14 días. La duración del tratamiento dependerá de la severidad y localización de la infección y de la evolución clínica y bacteriológica del paciente. Pacientes con compromiso renal: No es necesario ajustar la dosis de tigeciclina en pacientes con compromiso renal o en pacientes bajo hemodiálisis. Pacientes con compromiso hepático: No es necesario ajustar la dosis en pacientes con compromiso hepático leve a moderado (Child Pugh A y Child Pugh B). En base al perfil farmacocinético de tigeciclina en pacientes con compromiso hepático severo (Child Pugh C), deberá modificarse la dosis de tigeciclina a 100 mg seguida de 25 mg cada 12 horas. Se recomienda precaución y monitoreo de la respuesta al tratamiento en pacientes con insuficiencia hepática severa (Child Pugh C). Nińos: No se han establecido la seguridad y la eficacia de la tigeciclina en pacientes menores de 18 ańos y, por lo tanto, no se recomienda su administración en estos pacientes. (ver sección Advertencias y precauciones especiales para su uso). Ancianos: No se requieren ajustes posológicos en los ancianos. Raza y sexo: No se requieren ajustes posológicos basados en la raza o el sexo. Forma de administración: Infusión endovenosa.
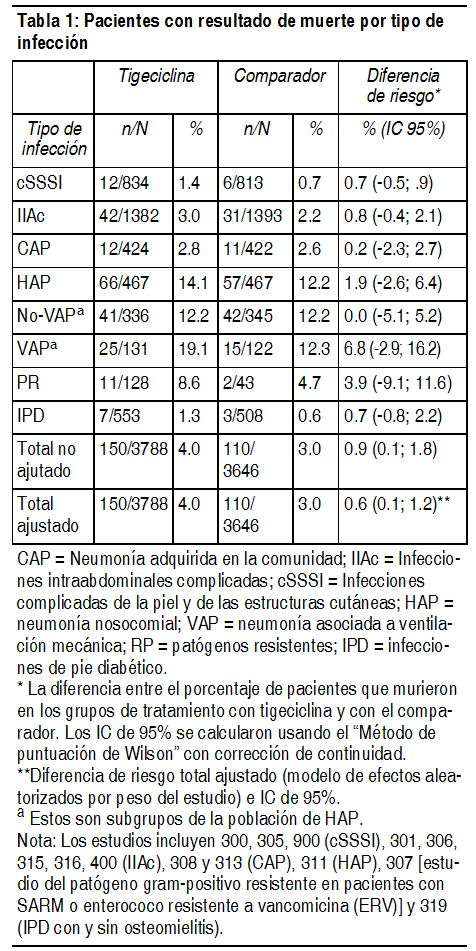
Efectos Colaterales:La frecuencia esperada de las reacciones adversas se presenta en categorías de frecuencia de CIOMS:Muy frecuentes: ≥10%. Frecuentes: ≥1% y <10%. Poco frecuentes: ≥0.1% y ≤1%. Raras: ≥0.01% y ≤0.1%. Muy poco frecuentes: ≤0.01%. En los pacientes que recibieron tigeciclina, se informaron las siguientes reacciones adversas: Clase de sistema orgánico/Reacción adversa: Trastornos de la sangre y el sistema linfático:Frecuentes: Tiempo parcial de tromboplastina activada (TPTA) prolongado, tiempo de protrombina (TP) prolongado. Trombocitopenia. Infrecuentes: Aumento del índice internacional normalizado (INR). Trastornos del sistema Inmune:Frecuencia no determinada: Reacciones anafilácticas/anafilactoideas. Trastornos metabólicos y nutricionales:Frecuentes: Bilirrubinemia, elevación del nitrógeno ureico en sangre; hipoproteinemia; hipoglicemia. Trastornos del sistema nervioso:Frecuentes: Mareos. Trastornos cardíacos:Frecuentes: Flebitis. Infrecuentes: Tromboflebitis. Trastornos respiratorios, torácicos y mediastínicos: Frecuentes: Pneumonía. Trastornos gastrointestinales:Muy frecuentes: Náuseas, vómitos, diarrea. Frecuentes: Anorexia, dolor abdominal, dispepsia. Infrecuentes: Pancreatitis aguda. Trastornos hepatobiliares: Frecuentes: Elevación de aspartato aminotransferasa (AST) en suero, elevación de alanina aminotransferasa (ALT) en suero* Infrecuentes: Ictericia. Frecuencia no determinada: Colestasis hepática. * En los pacientes tratados con tigeciclina, las anormalidades de AST y ALT se comunicaron con mayor frecuencia en el período postratamiento, mientras que en los grupos tratados con comparador, se informaron con mayor frecuencia durante el tratamiento. Trastornos del tejido cutáneo y subcutáneo:Frecuentes: Prurito, exantema; reacciones cutáneas severas, incluyendo síndrome de Stevens-Johnson. Trastornos generales y en el sitio de la administración: Frecuentes: Cefalea; Curación anormal, reacción en el sitio de la inyección. Infrecuentes: Flebitis, edema, dolor, inflamación en el sitio de la inyección. Pruebas complementarias:Frecuentes: Elevación de amilasa sérica. En un análisis en conjunto de los 13 estudios de fases 3 y 4 que incluyeron un comparador, la muerte ocurrió en el 4.0% (150/3788) de los pacientes que recibieron tigeciclina y en el 3.0% (110/3646) de los pacientes que recibieron medicamentos comparadores. En un análisis en conjunto de estos estudios, la diferencia de riesgo de la mortalidad por todas las causas fue 0.9% (IC de 95% 0.1, 1.8) entre los pacientes tratados con tigeciclina y los tratados con un comparador. En un análisis en conjunto de estos estudios, con base en un modelo de efectos aleatorizados por peso de estudio, existió una diferencia de riesgo ajustado del 0.6% (IC de 95% 0.1, 1.2) entre los pacientes tratados con tigeciclina y los que fueron tratados con un comparador. No se observaron diferencias significativas entre tigeciclina y los comparadores en cada tipo de infección (ver Tabla 1). La causa del desequilibrio no fue establecida. Generalmente, las muertes fueron el resultado de un empeoramiento de la infección o de complicaciones de la infección o de comorbilidades subyacentes.
Las reacciones adversas que surgieron más comúnmente en pacientes tratados con tigeciclina fueron náuseas 29.9% (19.3% leves; 9.2% moderadas; 1.4% severas) y vómitos 19.9% (12.1% leves; 6.8% moderadas; 1.1% severas). En general, náuseas o vómitos ocurrieron al inicio del tratamiento (días 1-2). La discontinuación de tigeciclina estuvo mayoritariamente asociada con náuseas (1.6%) y vómitos (1.3%).
Contraindicaciones: El uso de tigeciclina está contraindicado en pacientes con hipersensibilidad conocida a la tigeciclina
Advertencias: Los antibióticos de la clase de glicilciclina son estructuralmente similares a los antibióticos de la clase de tetraciclina. Por lo tanto, tigeciclina debería ser administrada con cuidado en pacientes con hipersensibilidad conocida a los antibióticos de la clase de tetraciclina. Los resultados de los estudios realizados en ratas con tigeciclina mostraron coloración anormal ósea. Tigeciclina puede asociarse con una coloración anormal permanente en los dientes de los seres humanos durante la dentición. Se observó un aumento en la mortalidad por todas las causas, en los estudios clínicos de fase 3 y 4 en pacientes tratados con tigeciclina en comparación con los pacientes tratados con comparador. En un análisis de los 13 estudios clínicos de fase 3 y 4 que incluyeron un comparador, la muerte ocurrió en el 4.0% (150/3788) de los pacientes que recibieron tigeciclina y en el 3.0% (110/3646) de los pacientes que recibieron medicamentos comparadores lo que se tradujo en una diferencia de riesgo sin ajustar del 0.9% (IC del 95% 0.1, 1.8). En un análisis en conjunto de estos estudios, con base en un modelo de efectos aleatorizados por peso de estudio, existió una diferencia de riesgo ajustado del 0.6% (IC de 95% 0.1, 1.2) entre los pacientes tratados con tigeciclina y los que fueron tratados con un comparador. La causa de este aumento no fue establecida. Este aumento de la mortalidad por todas las causas debería considerarse cuando se haga una selección entre las opciones de tratamiento (ver sección Efectos colaterales). Las reacciones de anafilaxia/anafilactoides fueron informadas con casi todos los agentes antibacterianos, incluso con tigeciclina y pueden poner en riesgo la vida. Se ha informado colitis pseudomembranosa con casi todos los agentes antibacterianos y la misma puede variar en gravedad desde leve hasta potencialmente mortal. Por lo tanto, es importante tener en cuenta este diagnóstico en pacientes que presentan diarrea con posterioridad a la administración de algún agente antibacteriano. Se debe tomar precaución cuando se considera monoterapia con tigeciclina en pacientes con infecciones intra-abdominales complicadas (IIAc) secundarias a una aparente perforación intestinal clínica. Los estudios en fase 3 y 4 de IIAc (n=2775), 140/1382 pacientes tratados con tigeciclina y 142/1393 pacientes tratados con comparadores presentaron perforaciones intestinales. De estos pacientes, 8/140 pacientes tratados con tigeciclina y 8/142 pacientes tratados con comparadores desarrollaron sepsis/ shock séptico. La relación del resultado con el tratamiento no se pudo establecer. Se han informado casos aislados de disfunción hepática significativa y deterioro hepático en pacientes que estaban siendo tratados con tigeciclina. Los antibióticos de la clase de glicilciclina son estructuralmente similares a los antibióticos de la clase de tetraciclina y pueden tener efectos adversos similares. Dichos efectos pueden incluir: Fotosensibilidad, pseudotumor cerebral, pancreatitis y acción antianabólica (que llevó a un aumento del BUN, azoemia, acidosis e hiperfosfatemia). Hubo casos (poco frecuentes) de pancreatitis aguda, que puede ser mortal, en asociación con el tratamiento de tigeciclina (ver sección Efectos colaterales). El diagnóstico de pancreatitis aguda debe tenerse en cuenta en pacientes que toman tigeciclina y desarrollaron síntomas clínicos, signos, o anomalías de laboratorio que sugieran pancreatitis aguda. Se han informado casos en pacientes sin factores de riesgo conocidos para pancreatitis. Los pacientes generalmente mejoran luego de la suspensión del tratamiento con tigeciclina. Debe analizarse la posibilidad de suspensión del tratamiento con tigeciclina en caso de que se sospeche que el paciente ha desarrollado pancreatitis. No se han establecido la seguridad ni la eficacia de tigeciclina en pacientes con neumonía nosocomial. En un estudio de pacientes con neumonía nosocomial, los pacientes fueron divididos aleatoriamente para recibir tigeciclina (100 mg inicialmente, luego 50 mg cada 12 horas) o un comparador. Asimismo, se permitió que los pacientes recibieran tratamientos específicos adyuvantes. El subgrupo de pacientes con neumonía asociada a ventilación mecánica que recibió tigeciclina tuvo tasas de curación menores (47.9% en comparación con 70.1% para la población clínicamente evaluable) y mayor mortalidad (25/131 [19.1%] en comparación con 15/122 [12.3%]) que el comparador. De esos pacientes con neumonía asociada a ventilación mecánica y bacteriemia en el período inicial, los que recibieron tigeciclina tuvieron una mortalidad mayor (9/18 [50.0%] en comparación con 1/13 [7.7%]) que el comparador. Al igual que con otras preparaciones antibióticas, el uso de este medicamento puede provocar un crecimiento excesivo de organismos no susceptibles, incluidos los hongos. Los pacientes deben ser cuidadosamente monitoreados durante el tratamiento. De ocurrir una sobreinfección, deben tomarse las medidas adecuadas.
Precauciones:Embarazo: Tigeciclina puede causar dańo fetal si se administra a una mujer embarazada. Los resultados de los estudios en animales indican que tigeciclina cruza la placenta y se encuentra en tejidos fetales. Con tigeciclina, se observó la disminución del peso fetal en ratas y conejos (con retrasos en la osificación asociados) y muerte fetal en conejos. Tigeciclina no fue teratogénica en las ratas ni en los conejos. En estudios preclínicos sobre seguridad, tigeciclina marcada con 14C cruzó la placenta y se encontró en los tejidos fetales, incluso en las estructuras óseas fetales. La administración de tigeciclina se asoció con ligeras reducciones en los pesos fetales y con una mayor incidencia de anomalías esqueléticas menores (retrasos en la osificación) con exposiciones equivalentes a 4.7 veces y a 1.1 veces la dosis diaria humana basada en el ABC en ratas y conejos, respectivamente. Se observó una mayor incidencia de muerte fetal con exposiciones equivalentes a 1.1 veces la dosis diaria humana basada en el ABC en conejos, en dosificaciones que producen una toxicidad materna mínima. No existen estudios de tigeciclina adecuados y bien controlados en mujeres embarazadas. Tigeciclina se debe usar durante el embarazo solamente si los beneficios potenciales justifican el riesgo potencial para el feto. No se ha estudiado el uso de tigeciclina durante el trabajo de parto y el parto. Lactancia: Los resultados de estudios en animales con el uso de tigeciclina marcada con 14C indican que tigeciclina se excreta en la leche de las ratas en período de lactancia. En concordancia con la biodisponibilidad oral limitada de tigeciclina, la exposición a tigeciclina en las crías lactantes a consecuencia de la exposición mediante la lecha materna es escasa o nula. Se desconoce si este medicamento se excreta en la leche humana. Debido a que muchos medicamentos se excretan en la leche humana, se debe tener precaución cuando se administra tigeciclina a las mujeres en período de lactancia (ver sección Advertencias y precauciones especiales para su uso).
Interacciones Medicamentosas: Tigeciclina (100 mg seguidos por 50 mg cada 12 horas) y digoxina (0.5 mg seguidos por 0.25 mg cada 24 horas) fueron coadministradas a sujetos sanos en un estudio de interacción medicamentosa. Tigeciclina disminuyó ligeramente la Cmáx. de digoxina en un 13%, pero no afectó el ABC ni la depuración de digoxina. Este pequeńo cambio en la Cmáx. no afectó los efectos farmacodinámicos de la fase estable de digoxina, medida por los cambios en los intervalos del ECG. Asimismo, digoxina no afectó el perfil farmacocinético de tigeciclina. Por lo tanto, no es necesario ajustar la dosis cuando tigeciclina se administra junto con digoxina. La administración concomitante de tigeciclina (100 mg seguidos por 50 mg cada 12 horas) y warfarina (dosis única de 25 mg), en sujetos sanos, provocó una disminución en la depuración de R-warfarina y S-warfarina en 40% y 23%; así como también una disminución en el ABC en un 68% y un 29%, respectivamente. Tigeciclina no modificó significativamente los efectos de warfarina sobre el Índice Normalizado Internacional (INR) aumentado. Asimismo, warfarina no afectó el perfil farmacocinético de tigeciclina. Sin embargo, si se administran tigeciclina y warfarina juntas, debe monitorearse el tiempo de protrombina o cualquier otra prueba de anticoagulación adecuada. Los estudios in vitro en microsomas hepáticos humanos indican que tigeciclina no inhibe el metabolismo mediado por ninguna de las 6 isoformas del citocromo CYP450: 1A2, 2C8, 2C9, 2C19, 2D6 y 3A4. Por lo tanto, no se espera que tigeciclina modifique el metabolismo de los medicamentos metabolizados por estas enzimas. Asimismo, dado que tigeciclina no se metaboliza extensivamente, no se espera que la depuración de tigeciclina se vea afectada por medicamentos que inhiban o induzcan la actividad de estas isoformas CYP450. Estudios in vitro que utilizan células Caco-2 indican que tigeciclina no inhibe el flujo de digoxina, lo que sugiere que tigeciclina no es un inhibidor de la glicoproteína P (P-gp). Esta información in vitro es consistente con la falta del efecto de tigeciclina en la depuración de digoxina, demostrada en el estudio de la interacción in vivo de la droga descrita más abajo. Tigeciclina es una sustrato de P-gp basada en un estudio in vitro que utiliza una línea celular que sobreexpresa P-gp. El potencial de contribución del transporte mediado por P-gp para la disponibilidad in vivo de tigeciclina se desconoce. La coadminstración de los inhibidores de P-gp (por ejemplo ketoconazol o ciclosporina) o inductores P-gp (por ejemplo, rifampicina) puede afectar la farmacocinética de tigeciclina. El uso concomitante de los antibióticos con anticonceptivos orales puede disminuir la efectividad de los anticonceptivos. Interferencias con pruebas de laboratorio y otras pruebas de diagnóstico: No se informó la existencia de interacciones entre las pruebas de laboratorio y los medicamentos.
Sobredosificación: No hay información específica disponible sobre el tratamiento de sobredosis con tigeciclina. La administración intravenosa de tigeciclina en una dosis única de 300 mg durante 60 minutos en voluntarios sanos provocó una mayor incidencia de las náuseas y los vómitos. En estudios de toxicidad con dosis única IV llevados a cabo con tigeciclina en ratones, la mediana de la dosis letal (LD50) fue 124 mg/kg en los machos y 98 mg/kg en las hembras. En ratas, la LD50 estimada fue 106 mg/kg para ambos sexos. La hemodiálisis no elimina tigeciclina en cantidades significativas. Carcinogenicidad: No se realizaron estudios durante todo el ciclo vital en animales para evaluar la carcinogenicidad potencial de la tigeciclina. Mutagenicidad: No se encontró potencial mutagénico ni clastogénico en una serie de pruebas, incluido un ensayo in vitro de aberración cromosómica en células de ovario de hámster chino (CHO), un ensayo de mutación primaria in vitro en células de CHO (locus HGRPT), ensayos in vitro de mutación primaria en células de linfoma de ratón y un ensayo micronúcleo in vivo. Deterioro de la fertilidad: Tigeciclina no afectó el apareamiento ni la fertilidad en ratas con exposiciones de hasta 4.7 veces la dosis diaria humana según el ABC. En las ratas hembra, no hubo efectos relacionados con el compuesto sobre los ovarios o los ciclos estro con exposiciones de hasta 4.7 veces la dosis diaria humana según el ABC. Otros: Se observó una disminución en los eritrocitos, los reticulocitos, los leucocitos y las plaquetas, en asociación con hipocelularidad de la médula ósea con exposiciones a tigeciclina de 8.1 veces y 9.8 veces la dosis diaria humana con base en el ABC en ratas y perros, respectivamente. Se comprobó que estas alteraciones se revierten dos semanas después de la dosis. La administración en bolo intravenoso de tigeciclina se ha asociado con una respuesta histaminica en estudios preclínicos. Estos efectos se observaron con exposiciones de 14.3 y 2.8 veces la dosis diaria humana con base en el ABC en ratas y perros, respectivamente. No se observó evidencia de fotosensibilidad en ratas luego de la administración de tigeciclina.
 Laboratorio
Laboratorio