Composición: Cada cįpsula contiene: 12.5; 25; 37.5 ó 50 mg de Sunitinib (como maleato). Excipientes: Sutent cįpsulas 12.5 mg:Contenido de la cįpsula: Manitol, Croscarmelosa Sódica, Povidona (K25), Estearato de Magnesio. Composición de la cįpsula: Gelatina, Óxido de Hierro Rojo y Dióxido de Titanio. Composición de la tinta de impresión: Shellac, Alcohol Deshidratado, Alcohol Isopropķlico, Alcohol Butķlico, Propilenglicol, Hidróxido de Sodio, Povidona, Dióxido de Titanio. Sutent cįpsulas 25 mg:Contenido de la cįpsula: Manitol, Croscarmelosa Sódica, Povidona (K25), Estearato de Magnesio. Composición de la cįpsula: Gelatina, Óxido de Hierro Negro, Óxido de Hierro Rojo, Óxido de Hierro Amarillo y Dióxido de Titanio. Composición de la tinta de impresión: Shellac, Alcohol Deshidratado, Alcohol Isopropķlico, Alcohol Butķlico, Propilenglicol, Hidróxido de Sodio, Povidona, Dióxido de Titanio. Sutent cįpsulas 50 mg:Contenido de la cįpsula: Manitol, Croscarmelosa Sódica, Povidona (K25), Estearato de Magnesio. Composición de la cįpsula: Gelatina, Óxido de Hierro Negro, Óxido de Hierro Rojo, Óxido de Hierro Amarillo y Dióxido de Titanio. Composición de la tinta de impresión: Shellac, Alcohol Dehidratado, Alcohol Isopropķlico, Alcohol Butķlico, Propilenglicol, Hidróxido de Sodio, Povidona, Dióxido de Titanio.
Indicaciones:Tumores del estroma gastrointestinal (GIST): Sutent estį indicado en el tratamiento de los tumores del estroma gastrointestinal (GIST), en adultos después del fracaso del tratamiento con mesilato de imatinib debido a resistencia o intolerancia. La experiencia con Sutent como tratamiento de primera lķnea es limitada. Carcinoma metastįsico de células renales (CMCR): Sutent estį indicado en el tratamiento del carcinoma metastįsico avanzado de células renales (CMCR) en adultos. Tumor pancréatico neuroendrocrino (pNET): Sutent estį indicado para el tratamiento de tumores pancreįticos neuroendocrinos (pNET) bien diferenciados, no extraķbles por cirugķa o metastįticos, con progresión de la enfermedad en adultos.
Posologķa: La terapia con sunitinib deberķa ser iniciada por un médico con experiencia en la administración de agentes anticancerķgenos. Para el tratamiento de carcinoma metastįsico de células renales (CMCR) y tumores del estroma gastrointestinal (GIST); la dosis recomendada de Sutent, es de 50 mg tomada oralmente todos los dķas, durante 4 semanas consecutivas, seguidas por un perķodo sin tratamiento de 2 semanas (Régimen 4/2), para abarcar un ciclo completo de 6 semanas. Para el tratamiento de tumores pancreaticos neuroendocrinos (pNET) la dosis recomendada de Sutent es de 37.5 mg por dķa de manera continua. Ajustes de la dosis:Seguridad y tolerabilidad: Para el tratamiento de carcinoma metastįsico de células renales (CMCR) y tumores del estroma gastrointestinal (GIST); las dosis se pueden modificar, segśn la seguridad y tolerabilidad individual, en incrementos de 12.5 mg. La dosis diaria no debe superar los 75 mg ni reducirse a menos de 25 mg. Para el tratamiento de tumores pancreįticos neuroendocrinos (pNET); las dosis se pueden modificar, segśn la seguridad y tolerabilidad individual, en incrementos de 12.5 mg. La dosis mįxima administrada en estudios fase 2 fue de 50 mg diarios. La interrupción de la dosis puede ser necesaria basada en la seguridad individual y la tolerabilidad. Inductores / inhibidores de CYP3A4: La coadministración del sunitinib con inductores potentes de la CYP3A4, tales como la rifampicina, deberķa evitarse (ver las secciones Advertencias e Interaciones Medicamentosas). Si esto no es posible, podrķa ser necesario aumentar la dosis del sunitinib, en incrementos de 12.5 mg, hasta un mįximo de 87.5 mg diarios para GIST y CMCR o 62.5 mg por dķa para pNET, sobre la base de un monitoreo cuidadoso de la tolerabilidad. La coadministración del sunitinib con inhibidores potentes de la CYP3A4, tales como ketoconazol, debe ser evitada. Si esto no es posible, podrķa ser necesario disminuir la dosis del sunitinib en 12.5 mg hasta llegar a un mķnimo de 37.5 mg diarios para el tratamiento de carcinoma metastįsico de células renales (CMCR) y tumores del estroma gastrointestinal (GIST) y un mķnimo de 25 mg diarios en el caso de tratamiento de tumores pancreįticos neuroendocrinos (pNET), sobre la base de un monitoreo cuidadoso de la tolerabilidad. Se recomienda seleccionar una medicación concomitante alternativa, con mķnimo o ningśn potencial para inducir o inhibir la CYP3A4. Uso en pediatrķa: No se han establecido la seguridad y la eficacia de sunitinib en pacientes pediįtricos (< 18 ańos). No hay información disponible. No hay uso relevante de Sutent en nińos desde el nacimiento a menos de 6 ańos para la indicación de tumor no extirpable y/o tumor del estroma gastrointestinal metastįsico maligno después de la falla del tratamiento con imatinib mesilato debido a resistencia o intolerancia. No hay uso relacionado de Sutent en población pediįtrica en las indicaciones de carcinoma de células renales avanzado/metastįtico y tumores neuroendocrinos pancreįticos bien diferenciados con progresión de la enfermedad. El uso de Sutent en población pediįtrica no estį recomendado. Uso en pacientes de edad avanzada (ancianos) (≥ 65 ańos de edad): Aproximadamente 1/3 de los sujetos participantes en los estudios clķnicos de sunitinib, tenķan 65 o mįs ańos de edad. No se observaron diferencias significativas en seguridad o eficacia entre los pacientes jóvenes y mayores. Insuficiencia hepįtica: No es necesario ajustar la dosis cuando se inicia el tratamiento con Sutent en pacientes con insuficiencia hepįtica leve (Clase A Child-Pugh) o moderada (Clase B Child-Pugh). Sunitinib no ha sido estudiado en sujetos con insuficiencia hepįtica severa (Clase C Child-Pugh). Insuficiencia renal: No es necesario ajustar la dosis cuando se inicia el tratamiento con Sutent en pacientes con insuficiencia renal (leve-severa) o en enfermedad renal terminal (ESRD) en hemodiįlisis. Los ajustes de dosis posteriores deberķan basarse en la seguridad y tolerabilidad individual del paciente. Método de administración: Sunitinib es para administración oral. Se puede tomar con o sin alimentos. Si se olvida una dosis, el paciente no debe tomarse una dosis adicional. El paciente debe tomarse la dosis usual prescrita el dķa siguiente.
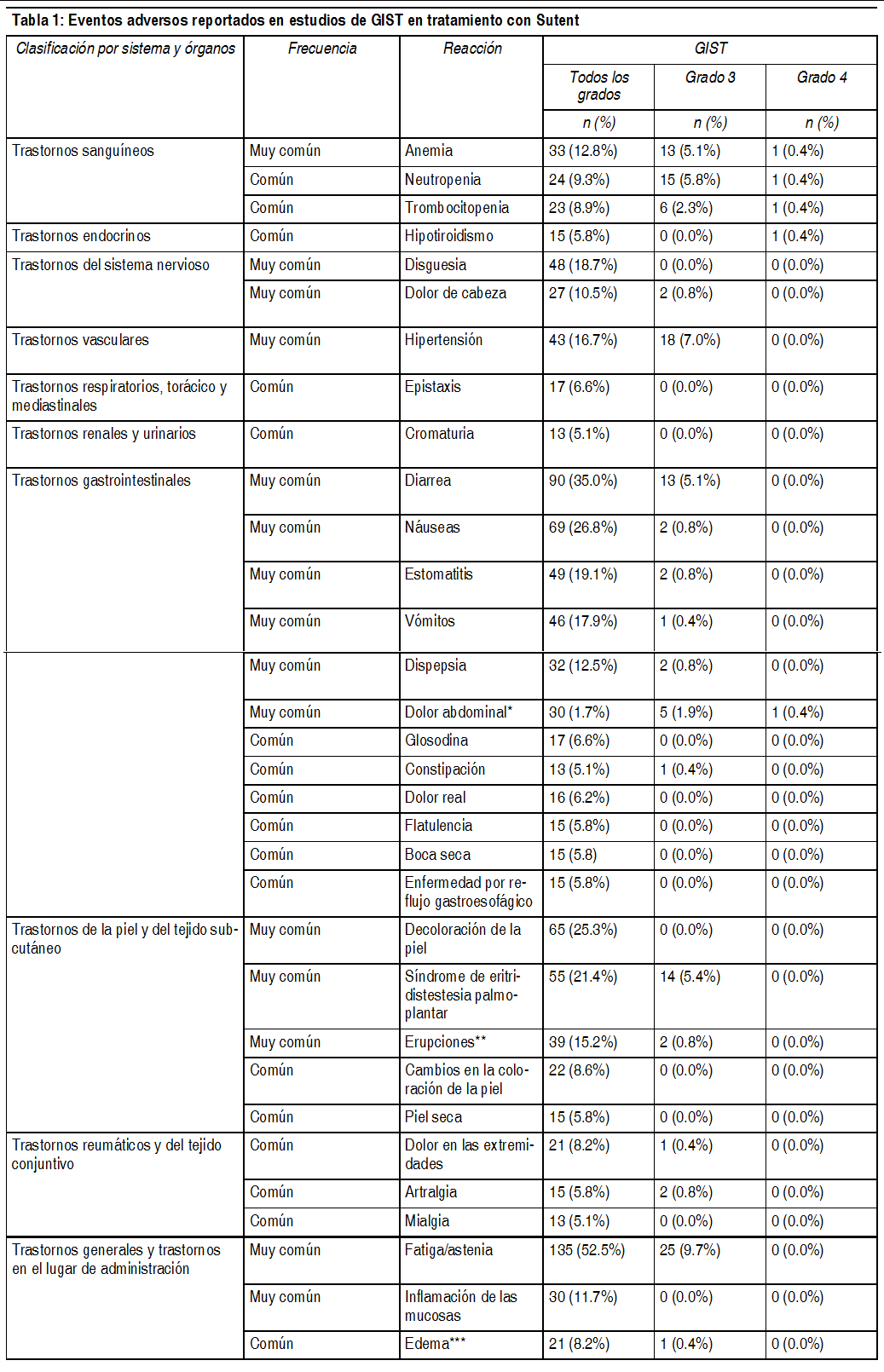
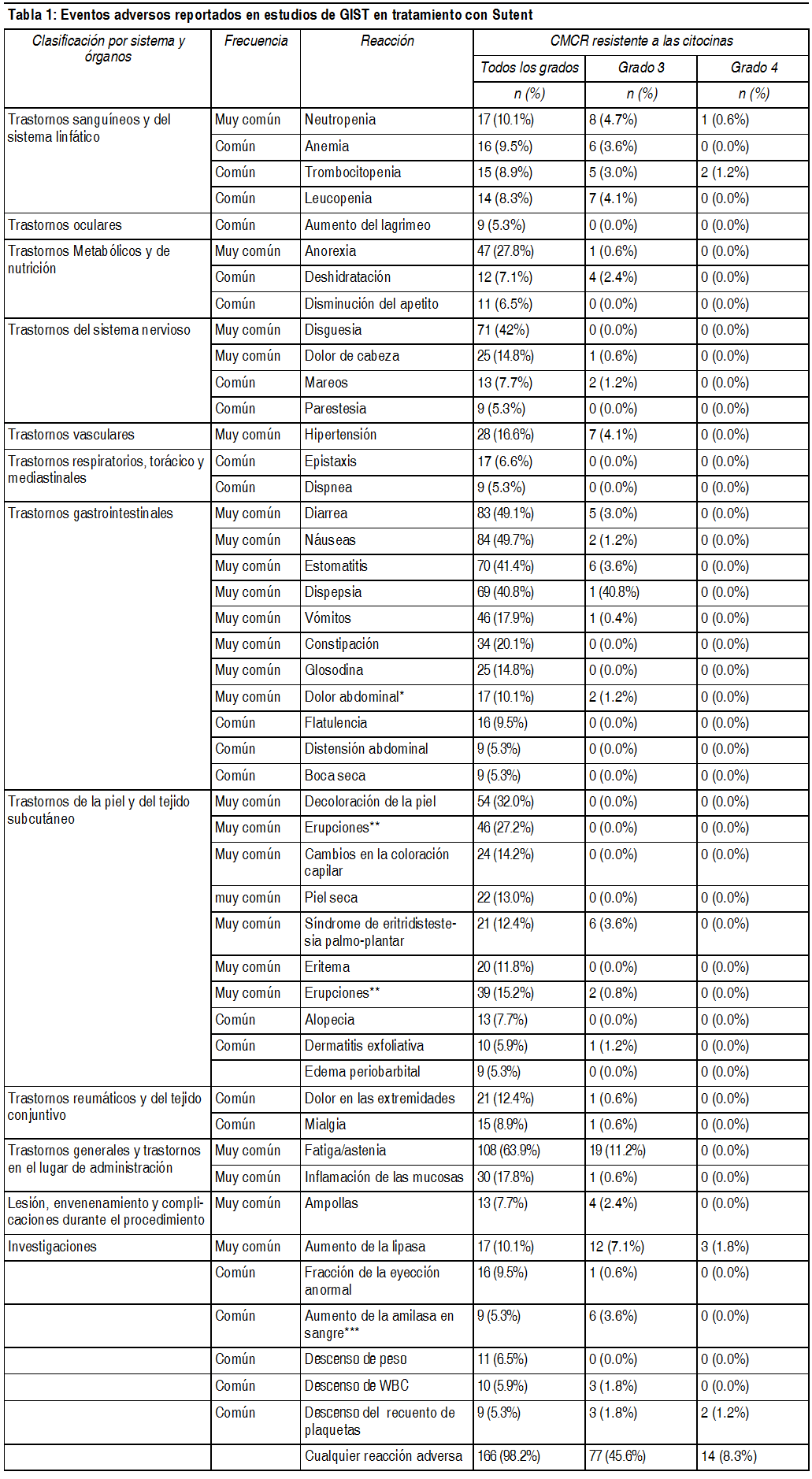
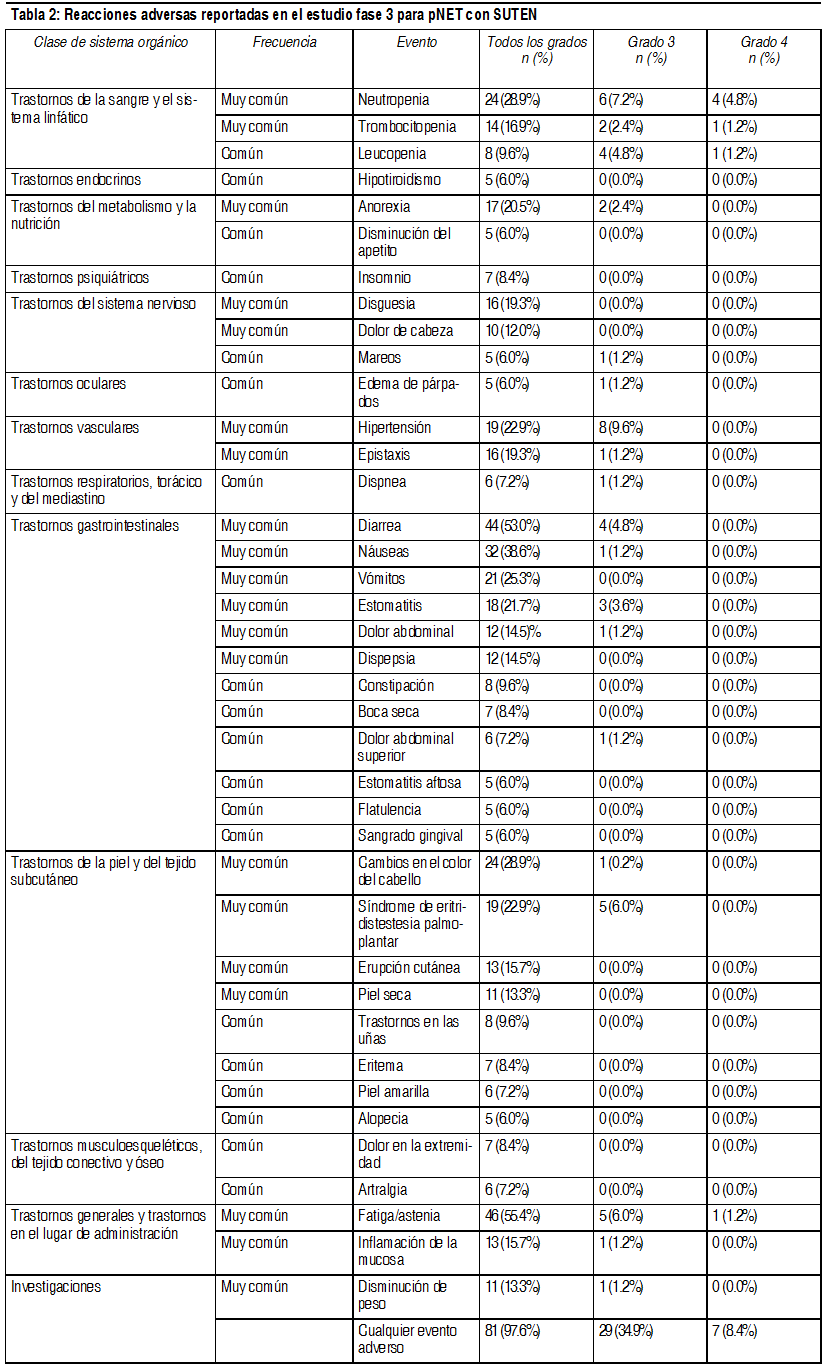
Efectos Colaterales: Los eventos adversos graves mįs importantes relacionados con Sutent en el tratamiento, de pacientes con tumores sólidos fueron, embolismo pulmonar (1%), trombocitopenia (1%), hemorragia tumoral (0.9%), neutropenia febril (0.4%) e hipertensión (0.4%). Los eventos adversos mįs comunes relacionados con el tratamiento (experimentado al menos por el 20% de los pacientes), de cualquier grado, incluyeron: fatiga; trastornos gastrointestinales, tales como diarrea, nįuseas, estomatitis, dispepsia y vómitos; decoloración de la piel; disgeusia y anorexia. Fatiga, hipertensión y neutropenia, fueron los eventos adversos mįs comunes relacionados con el tratamiento, con una severidad mįxima de Grado 3, y lipasa aumentada fue el evento adverso mįs frecuente relacionado con el tratamiento con una gravedad mįxima de Grado 4 en pacientes con tumores sólidos. La epistaxis relacionada con el tratamiento, fue la reacción adversa informada con mayor frecuencia, se presentó en el 8% de pacientes con tumores sólidos. En aproximadamente la mitad de los pacientes con tumores sólidos que experimentaron reacciones hemorrįgicas (consulte también la sección Advertencias). En estudios clķnicos de sunitinib, se observaron convulsiones en sujetos con evidencia radiológica de metįstasis cerebral. Ademįs, ha habido informes muy poco frecuentes (<1%), algunos fatales, de sujetos que sufrieron convulsiones y, posteriormente, evidencia radiológica de la presencia del sķndrome de leucoencefalopatķa posterior reversible (SLPR). Las frecuencias estįn definidas como: muy comśn (≥1/10), comśn (≥1/100 a < 1/10), poco comśn (≥1/1000 a <1/100), raro (≥1/10000 a < 1/1000), muy raro (<1/10000), desconocido (no puede estimarse de la información disponible).
Se han informado las siguientes reacciones adversas adicionales en estudios clķnicos con sunitinib. Trastornos cardķacos: Poco frecuentes: Insuficiencia cardķaca, insuficiencia cardķaca congestiva, insuficiencia del ventrķculo izquierdo. Raras: intervalo QT prolongado, torsade de pointes. Trastornos gastrointestinales: Poco frecuentes: pancreatitis. Raras: perforación gastrointestinal. Trastornos hepatobiliares: Poco frecuentes: Insuficiencia hepįtica, colecistitis, especialmente colecistitis acalculosa. Trastornos de la piel y tejido subcutįneo: Raro: Sķndrome de Stevens-Johnson. Investigaciones frecuentes: Hormona estimulante de la tiroides (TSH, por sus siglas en inglés) elevada, aumento del įcido śrico en sangre. Experiencia post-marketing: Las siguientes reacciones adversas han sido identificadas durante el uso de post-aprobación de sunitinib. Dado que estas reacciones son reportadas voluntariamente por una población de tamańo incierto, no siempre es posible estimar de manera fiable su frecuencia o establecer una relación causal con la exposición al fįrmaco. Infecciones: Casos de infecciones serias (con o sin neutropenia), incluyendo neumonia, han sido reportados. Algunos casos con desenlace fatal. Las infecciones mįs comśnmente observados con el tratamiento con sunitinib son las infecciones tķpicamente observadas en pacientes con cįncer, por ejemplo, las vķas respiratorias, tracto urinario, infecciones de la piel y sepsis. Se han reportado casos raros de fascitis necrotizante, entre ellos del perineo, a veces mortal. Trastornos sanguķneos y del sistema linfįtico: Se han reportado raros casos de microangiopatķa trombótica. La suspensión temporal de Sutent es recomendada; después de su resolución, el tratamiento puede ser restaurado segśn el criterio del médico tratante. Trastornos del sistema inmune: Han sido reportadas reacciones de hipersensibilidad, incluyendo angioedema. Trastornos endocrinos: Raros casos de hipertiroidismo, algunos seguidos por hipotiroidismo, se han reportado en ensayos clķnicos y durante la experiencia post-comercialización. Poco frecuentes: tiroiditis. Desórdenes del sistema nervioso: Se ha reportado alteración del gusto, incluyendo ageusia. Desórdenes cardķacos: Se ha reportado cardiomiopatķa, en algunos casos con desenlace fatal. Eventos hemorrįgicos: Se han informado casos de hemorragia pulmonar, gastrointestinal, tumoral, del tracto urinario y cerebral, algunas de ellas fatales, en pacientes tratados con sunitinib. Trastornos hepatobiliares: Se ha reportado insuficiencia hepįtica y puede incluir anormalidades en las pruebas de función hepįtica, hepatitis o falla hepįtica. Trastornos gastrointestinales: Frecuentes: esofagitis. Trastornos musculoesqueléticos y del tejido conectivo: Raros: casos de miopatķa y/o rabdomiolisis, algunos con insuficiencia renal aguda, han sido reportados. Pacientes con signos o sķntomas de toxicidad muscular deberķan ser manejados de acuerdo a las prįcticas médicas estįndares. Se han reportado casos de formación de fķstula, veces asociadas con necrosis de tumor y regresión, en algunos casos con resultado fatal. Se han reportado problemas en la curación de heridas durante la terapia con Sutent. Se ha reportado casos de osteonecrosis de mandķbula (ONM) en pacientes tratados con Sutent, la mayorķa de estos pacientes tenķan factores de riesgo para ONM, en particular aquellos expuestos a bifosfonatos IV y/o con historia de enfermedad dental que requiere de procedimiento dental invasivo y/o estaban recibiendo medicación concomitante conocida de estar asociada con estas reacciones adversas. Trastornos renales y urinarios: Se han reportado casos de deterioro renal, falla renal y/o falla renal aguda, en algunos casos con resultado fatal. Se han reportado casos de proteinuria y raros casos de sķndrome nefrótico. Trastornos respiratorios: Se han reportado casos de embolismo pulmonar, algunos de ellos con resultado fatal. Desórdenes vasculares: Se han reportado casos de eventos tromboembólicos arteriales, a veces fatales, en pacientes tratados con sunitinib. Los eventos mįs comśnes fueron accidente cerebrovascular, ataque isquémico transitorio e infarto cerebral. Los factores de riesgo asociados con ATE, ademįs de la enfermedad subyacente maligna y 65 ańos de edad, incluidas hipertensión, diabetes mellitus y enfermedad tromboembólica previa. Desórdenes del metabolismo y la nutrición: Se han reportado casos de TLS (sķndrome de la lisis del tumor), algunos fatales, en pacientes tratados con sunitinib.
Contraindicaciones: El uso estį contraindicado en pacientes con hipersensibilidad al sunitinib o a alguno de los excipientes.
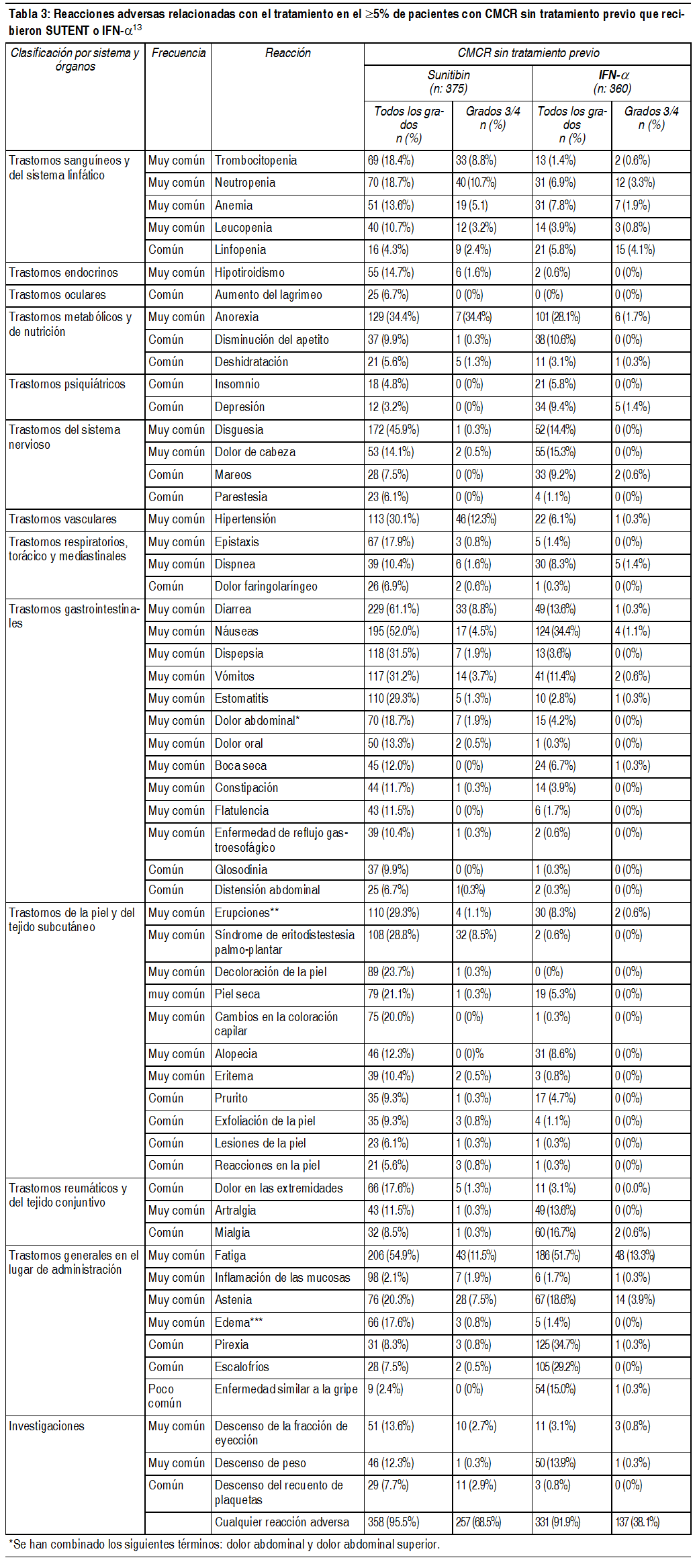
Advertencias:Trastornos de la piel y tejidos: La decoloración de la piel, posiblemente debido al color (amarillo) de la sustancia activa, es un evento adverso comśn, el cual ocurre en aproximadamente el 30% de los pacientes. Los pacientes deben ser advertidos de que podrķa ocurrir despigmentación del cabello o de la piel, durante el tratamiento con sunitinib. Otros efectos dermatológicos posibles incluyen resequedad, engrosamiento o agrietamiento de la piel, ampollas o erupción ocasional en las palmas de las manos y plantas de los pies. Los eventos antes mencionados no fueron acumulativos, tķpicamente fueron reversibles y generalmente no resultaron en la descontinuación del tratamiento. Se han reportado reacciones cutįneas severas, incluyendo casos de eritema multiforme (EM) y casos sugerentes de sķndrome de Stevens-Johnson (SSJ), algunos de los cuales fueron mortales. Si se presentan signos o sķntomas de SSJ o EM (ej. rash progresivo en la piel a menudo con ampollas o lesiones de la mucosa), el tratamiento con Sutent debe ser discontinuado. Si el diagnóstico de SSJ es confirmado, el tratamiento no debe reiniciarse. En algunos casos de EM sospechado, los pacientes toleran la reintroducción de la terapia de Sutent a dosis mįs bajas luego de resuelta la reacción; algunos de estos pacientes también reciben tratamiento concomitante con corticosteroides o antihistamķnicos. Eventos hemorrįgicos y sangrado de tumores. Eventos hemorrįgicos, algunos de ellos fatales, reportados a través de la experiencia post-comercialización han incluido hemorragias gastrointestinales, respiratorias, tumorales, del tracto urinario y cerebrales. En ensayos clķnicos, aproximadamente el 2% de los pacientes con GIST, presentaron hemorragia del tumor relacionada con el tratamiento. Estos eventos pueden ocurrir sśbitamente y en el caso de los tumores de pulmón, pueden presentarse como hemoptisis o hemorragia pulmonar grave, con riesgo vital. Se han observado casos de hemorragia pulmonar, algunos con resultados fatales en estudios clķnicos, tambien se han informado en el perķodo de post-comercialización en pacientes tratados con sunitinib para CMCR, GIST y cįncer pulmonar metastįsico de células pequeńas (NSCLC, por sus siglas en inglés). Sunitinib no estį aprobado para uso en pacientes con NSCLC. Eventos de sangrado ocurrieron en 18% de los pacientes que recibieron sunitinib en el estudio Fase 3 de GIST, comparado con 17% de los pacientes que recibieron placebo. En los pacientes que recibķan sunitinib para el tratamiento de CMCR (carcinoma de células renales metastįsico) no tratado previamente (naļve), el 39% de los pacientes tuvieron eventos de sangrado, comparado con 11% de los pacientes que recibķan interferón- (IFN- ). Once (3.1%) de los pacientes con sunitinib versus 1 (0.3%) de los pacientes en IFN- experimentaron eventos hemorrįgicos Grado 3 o mayores relacionados con el tratamiento. De los pacientes que recibķan sunitinib para CMCR refractario a citocina, 26% experimentó sangrado. Eventos de sangrado, excluyendo epixtasis, ocurrieron en 19% de los pacientes que recibieron sunitinib en el estudio para pNET en fase 3 comparado con 4% de los pacientes que recibieron placebo. Los exįmenes de rutina para evaluar este evento, deben incluir recuento sanguķneo completo y examen fķsico. La epistaxis es la reacción adversa hemorrįgica mįs comśn, siendo reportada por aproximadamente la mitad de los pacientes con tumores sólidos que experimentaron eventos hemorrįgicos. Algunos casos de epistaxis fueron severos, pero muy raramente fatales. Trastornos del tracto gastrointestinal: Nįuseas, diarrea, estomatitis, dispepsia y vómitos fueron los eventos adversos gastrointestinales mįs reportados (ver sección Efetos colaterales). El tratamiento de soporte para eventos adversos gastrointestinales que requieren tratamiento puede incluir antieméticos o antidiarreicos. Complicaciones gastrointestinales graves, algunas veces fatales, incluyendo perforación gastrointestinal, han ocurrido raramente en pacientes con cįnceres intrabdominales tratados con sunitinib. Hipertensión: Hipertensión relacionada con el tratamiento fue reportada en aproximadamente el 16% de los pacientes con tumores sólidos. La dosis de sunitinib se redujo o suspendió temporalmente en aproximadamente el 2.7% de esta población de pacientes. A ninguno de estos pacientes se le discontinuó permanentemente el tratamiento con sunitinib. Se observó hipertensión severa (sistólica >200 mmHg o diastólica >110 mmHg) en el 4.7% de los pacientes con tumores sólidos. Se reportó hipertensión relacionada con el tratamiento en aproximadamente el 30% de los pacientes recibiendo sunitinib para el tratamiento de CMCR no tratado previamente (naļve), comparado con el 6% de los pacientes recibiendo IFN- . Ocurrió hipertensión severa en el 12% de los pacientes tratados con sunitinib y en el 6% de los pacientes tratados con IFN-α, no antes tratados (naļve). En el estudio fase 3 para pNET, se reportó hipertensión relacionada al tratamiento en un 23% de los pacientes que recibieron sunitinib, comparado con un 4% de los pacientes que recibķan placebo. Ocurrió hipertensión severa en 10% de los pacientes con pNET que recibķan sunitinib y 3% de los pacientes con placebo. Los pacientes deben ser sometidos a una evaluación para hipertensión y controlados de la forma mįs apropiada. Se recomienda la suspensión temporal en pacientes con hipertensión severa, que no estén siendo controlados con manejo médico. El tratamiento se puede reanudar cuando la hipertensión esté controlada. Trastornos hematológicos: Recuentos absolutos de neutrófilos disminuidos, de gravedad Grado 3 y 4, fueron reportados en un 13.1% y 0.9% de los pacientes respectivamente. Recuentos de plaquetas disminuidos, de gravedad Grado 3 y 4, fueron reportados en un 4% y 0.5% de los pacientes respectivamente. Los eventos anteriores no fueron acumulativos, tķpicamente fueron reversibles y generalmente no resultaron en la discontinuación del tratamiento. En algunos casos de post-comercialización se ha reportado hemorragia asociada a trombocitopenia. Se deben realizar recuentos sanguķneos completos al principio de cada ciclo de tratamiento en los pacientes que reciben tratamiento con Sutent. Trastornos cardķacos: Mediante la experiencia post-comercialización, eventos cardiovasculares han sido reportados, incluyendo falla cardķaca, cardiomiopatķas y trastornos del miocardio, algunos de los cuales fueron fatales. En estudios clķnicos, disminuciones de 20% y valores por debajo del lķmite normal de la fracción de eyección ventricular izquierda (FEVI), ocurrieron en aproximadamente el 2% de los pacientes con GIST tratados con sunitinib, en el 4% de los pacientes con CMCR refractario a citocina y en el 2% de los pacientes tratados con placebo. Estos descensos en la FEVI no parecieran ser progresivos y frecuentemente mejoraron a medida que continuaba el tratamiento. En el estudio de CMCR no tratado previamente (naļve), el 27% y 15% de los pacientes con sunitinib e IFN- , respectivamente, tuvieron un valor de FEVI por debajo del lķmite inferior de la normalidad (Lower Limit of Normal-LLN). A 2 pacientes (<1%) que recibķan sunitinib, se les diagnosticó insuficiencia cardķaca congestiva (ICC). Los eventos adversos relacionados con el tratamiento de "insuficiencia cardķaca", "insuficiencia cardķaca congestiva" o "insuficiencia ventricular izquierda", se reportaron en 0.7% de los pacientes con tumores sólidos y en 1% de los pacientes tratados con placebo. En estudio de fase 3 para pNET, 1 paciente (1%) que recibió sunitinib presentó falla cardķaca relacionada con el tratamiento. Los pacientes que habķan presentado eventos cardķacos dentro de los 12 meses previos a la administración de sunitinib, tales como infarto del miocardio (incluyendo angina severa/ inestable), injerto de bypass de arteria coronaria/periférica, insuficiencia cardķaca congestiva (ICC) sintomįtica, accidente cerebrovascular o ataque isquémico transitorio o embolismo pulmonar, fueron excluidos de los estudios clķnicos de sunitinib. Se desconoce si los pacientes con esas condiciones concomitantes puedan estar en mayor riesgo de desarrollar disfunción ventricular izquierda relacionada con la droga. Los pacientes deben ser monitoreados cuidadosamente, para identificar signos y sķntomas clķnicos de ICC, especialmente aquellos con factores de riesgo cardķaco y/o historial de enfermedad coronaria. A los médicos se les aconseja sopesar este riesgo, contra los posibles beneficios del medicamento. También se debe considerar la realización de evaluaciones periódicas de la FEVI, mientras el paciente esté recibiendo sunitinib. En los pacientes sin factores de riesgo cardķaco, se debe considerar la realización de una evaluación basal de la fracción de eyección. Si se presentan manifestaciones clķnicas de ICC, se recomienda la descontinuación de sunitinib. La administración de sunitinib debe ser interrumpida y/o reducida en los pacientes sin evidencia clķnica de ICC, pero con una fracción de eyección <50% y >20% por debajo de la basal. Prolongación del intervalo QT: Se ha evidenciado que en concentraciones aproximadamente el doble de las terapéuticas sunitinib prolonga (corrección de Fridericia) el intervalo QTcF. No hubo pacientes con prolongación del intervalo QT/QTc mayor al grado 2 de los Criterios de Terminologķas Comunes para Eventos Adversos (CTCAE) v3.0. La prolongación del intervalo QT puede llevar a un mayor riesgo de arritmias ventriculares, incluyendo torsade de pointes. El torsade de pointes ha sido observado en <0.1% de los pacientes expuestos a sunitinib. Sunitinib se debe usar con precaución en los pacientes con antecedentes conocidos de prolongación del intervalo QT, en los pacientes que estén tomado antiarrķtmicos o en los pacientes con enfermedad cardķaca relevante pre existente, bradicardia o trastornos electrolķticos. El tratamiento concomitante con inhibidores potentes de la CYP3A4, debe ser limitado debido a posibles aumentos de las concentraciones plįsmaticas de sunitinib, debiéndose implementar con precaución y reducir la dosis de sunitinib. Eventos tromboembólicos venosos: En el estudio fase 3, doble ciego, para GIST, 7 pacientes (3%) recibiendo Sutent y ninguno con placebo experimentaron eventos tromboembólicos venosos: 5 de 7 fueron trombosis venosa profunda (TVP) Grado 3 y 2 fueron Grado 1 ó 2. Cuatro de estos 7 pacientes con GIST interrumpieron su tratamiento tras la primera observación de TVP. Trece pacientes (3%) recibiendo sunitinib sin tratamiento previo en el estudio para CMCR y 4 pacientes (2%) en los dos estudios para CMCR citoquina-refractario han reportado eventos tromboembólicos venosos. Nueve de estos pacientes tuvieron embolismos pulmonares, 1 fue Grado 2 y 8 fueron Grado 4. Ocho de estos pacientes tuvo una TVP, 1 con Grado 1, 2 con grado 2, 4 con grado 3 y 1 de Grado 4. En uno de los casos se interrumpió la administración de la dosis. En tratamiento naļve CMCR para pacientes recibiendo IFN-α, ocurrieron 6 (2%) eventos tromboembólicos venosos, 1 paciente (<1%) experimentó TVP Grado 3 y 5 pacientes (1%) tuvieron embolismos pulmonares, todos Grado 4. La presentación de embolia pulmonar relacionada con el tratamiento se informó en aproximadamente el 1.1% de pacientes con tumores sólidos que recibķan sunitinib. Ninguno de estos eventos resultó en que un paciente discontinuara el tratamiento con sunitinib, aunque en unos pocos casos o se redujo la dosis o bien hubo una demora temporal en el tratamiento. No se presentaron otros episodios de embolia pulmonar en estos pacientes luego de que se reanudó el tratamiento. Eventos tromboembólicos arteriales: Han sido reportados casos de eventos tromboembólicos arteriales (ETA), algunas veces fatales, en pacientes tratados con sunitinib. Los eventos mįs frecuentes incluyeron accidentes cerebrovasculares, ataques isquémicos transitorios e infarto cerebral. Los factores de riesgo asociados con ETA, sumado a la enfermedad maligna subyacente y la edad ≥ 65 ańos, incluyendo hipertensión, diabetes mellitus, y enfermedad tromboembólica previa. Eventos respiratorios: Se ha informado casos de embolia pulmonar, algunos de ellos con desenlace fatal. Disfunción tiroidea: Se recomienda la medición basal de la función tiroidea, en el caso de los pacientes con hipotiroidismo o hipertiroidismo, estos deberķan ser tratados antes de iniciar el tratamiento con sunitinib, segśn la prįctica médica estįndar. Durante el tratamiento con sunitinib, todos los pacientes deberķan ser cuidadosamente observados para la aparición de signos y/o sķntomas de disfuncion tiroidea cada 3 meses. Los pacientes con signos y sķntomas sugestivos de disfunción tiroidea, deberķan ser monitoreados con exįmenes de laboratorio de función tiroidea y se deberķan tratar de acuerdo con la prįctica médica estįndar. Se advirtió hipotiroidismo adquirido emergente del tratamiento en 4% de los pacientes con GIST que recibieron sunitinib versus un 1% de los que recibieron placebo. Se informó hipotiroidismo como reacción adversa en un 16% de los pacientes que recibieron Sutent en el estudio de CMCR. Adicionalmente fueron reportadas elevaciones de la TSH en 4 pacientes (2%) con CMCR citoquina refractario. En total, el 7% de la población con CMCR citoquina refractario tuvo evidencia clķnica o de laboratorio de hipotiroidismo emergente del tratamiento. En el estudio fase 3 para pNET fue reportado en 5 pacientes (6%) que recibieron sunitinib y en 1 paciente (1%) que recibió placebo. Raros casos de hipertiroidismo, algunos seguidos por hipotiroidismo, se han reportado en ensayos clķnicos y durante la experiencia post-comercialización. Pancreatitis: Se observaron aumentos en la actividad de lipasa y amilasa sérica en pacientes en tratamiento por tumores sólidos con sunitinib. El aumento en las actividades de lipasa fue transitorio y generalmente no acompańado de signos y sķntomas de pancreatitis en sujetos con tumores sólidos. La pancreatitis ha sido observada extraordinariamente (<1%) de los pacientes que recibieron sunitinib para GIST o CMCR. Se han reportado casos de eventos pancreįticos serios, algunos con resultado fatal. Si se presentan sķntomas de pancreatitis, los pacientes deberķan descontinuar el tratamiento con sunitinib y deben recibir cuidados de soporte apropiados. Hepatotoxicidad: Se ha observado hepatotoxicidad en pacientes tratados con sunitinib. Casos de falla hepįtica, algunos con resultado fatal, se observaron en <1% de los pacientes tratados con sunitinib. Monitorizar mediante pruebas de función hepįtica (alanino transaminasa [ALT], aspartato transaminasa [AST], niveles de bilirrubina), antes de comenzar el tratamiento, durante cada ciclo de tratamiento y como esté indicado clķnicamente. La administración de sunitinib debe interrumpirse en caso de reacciones adversas relacionadas con el hķgado de Grado 3 ó 4, y descontinuarse si no hay solución. Función renal: Se han reportado casos de deterioro renal, falla renal y/o falla renal aguda, en algunos casos con resultado fatal. Se han reportado casos de proteinuria y casos raros de sķndrome nefrótico. Se recomienda realizar un anįlisis de orina para linea basal, y los pacientes deberķan ser monitoreados por el desarrollo o empeoramiento de proteinuria. Sunitinib debe discontinuarse en los pacientes con sķndrome nefrótico. Fķstula: Se han informado casos de formación de fistulas, asociados a veces a necrosis o regresión del tumor. Algunos de los casos tuvieron un desenlace fatal. Problemas con la curación de heridas: Se han reportados casos de problemas con la curación de heridas durante la terapia con sunitinib. Se recomienda interrupción temporal de la terapia con sunitinib como medida de precaución en pacientes que serįn sometidos a intervención quirśrgica mayor. Hay experiencia clķnica limitada con respecto a la iniciación del tratamiento después de una intervención quirśrgica mayor. Por lo tanto, la decisión de retomar la terapia con sunitinib luego de la intervención quirśrgica mayor deberķa basarse en el juicio clķnico de recuperación de la cirugķa. Osteonecrosis de mandķbula (ONM): Se han reportado casos de ONM en estudios clķnicos y durante la post- comercialización en pacientes tratados con Sutent. La mayorķa de los casos ocurrieron en pacientes que recibieron tratamiento previo o concomitante con bifosfonatos IV, para los cuales ONM es un riesgo identificado. Por tanto, se deberķa tener cuidado cuando Sutent y bifosfonatos IV se usen simultįnea o secuencialmente. Procedimientos dentales invasivos son tambien un factor de riesgo identificado. Antes del tratamiento con Sutent, deberķa considerarse un examen y odontologķa preventiva apropiada. En pacientes que han recibido o estįn recibiendo bifosfonatos IV, deberķa evitarse procedimientos dentales invasivos si esposible. Hipersensibilidad/ angioedema: Se han informado reacciones de hipersensibilidad, entre ellas, angioedema. Trastornos del sistema nervioso: Se ha informado de trastornos del sentido del gusto, entre ellos, ageusia. Convulsiones: En los estudios clķnicos y en la experiencia post-comercialización de sunitinib, se observaron convulsiones en sujetos con o sin evidencia radiológica de metįstasis cerebrales. Adicionalmente, han habido pocos reportes (<1%) de sujetos que presentaron convulsiones y evidencia radiológica del sķndrome de leucoencefalopatķa posterior reversible (SLPR). Los pacientes con convulsiones y signos/sķntomas compatibles con SLPR, tales como hipertensión, cefalea, estado de alerta disminuido, funcionamiento mental alterado y pérdida visual, incluyendo ceguera cortical, deberķan controlarse con atención médica, incluyendo control de hipertensión. Se recomienda la suspensión temporal de sunitinib; el tratamiento se puede reiniciar a discreción del médico tratante después de la resolución del problema. Sķndrome de lisis tumoral (SLT): Se han observado con poca frecuencia casos de SLT, algunos fatales, en ensayos clķnicos y durante la experiencia posterior al ingreso al mercado en pacientes tratados con sunitinib. Los pacientes que suelen tener riesgo de SLT son aquellos con una gran carga tumoral previa al tratamiento. Estos pacientes deben ser monitoreados de cerca y tratados como se indique clķnicamente. Fasceitis necrotizante: Se han informado casos raros de fasceitis necrotizante, entre ellos del periné, a veces mortal. Se deberķa interrumpir el tratamiento con sunitinib en pacientes que desarrollan fasceitis necrotizante, y se deberķa iniciar, con prontitud, el tratamiento adecuado. Proteinuria: Se han informado casos de proteinuria, asķ como casos raros de sķndrome nefrótico. Se recomienda realizar anįlisis de orina basales y monitorizar a los pacientes a fin de detectar el desarrollo o el empeoramiento de la proteinuria. La seguridad del tratamiento continuado con sunitinib en pacientes con proteinuria moderada a grave no se ha evaluado sistemįticamente. Se debe interrumpir sunitinib en pacientes con sķndrome nefrótico.
Precauciones:Riesgo de toxicidad: En los estudios de toxicidad de dosis repetidas en ratas y monos, de hasta 9 meses de duración, los efectos en los órganos blancos principales se identificaron en el tracto gastrointestinal (emesis y diarrea en monos), glįndula suprarrenal (congestión cortical y/o hemorragia en ratas y monos, con necrosis seguida por fibrosis en las ratas), sistema hemolinfopoyético (hipocelularidad de la médula ósea y depleción linfoide del timo, bazo y ganglios linfįticos), pįncreas exocrino (desgranulación de las células acinares con necrosis celular simple), glįndula salival (hipertrofia acinar), articulación ósea (engrosamiento de la placa de crecimiento), śtero (atrofia) y ovarios (desarrollo folicular disminuido). Todos los hallazgos ocurrieron en niveles de exposiciones plasmįticas de sunitinib clķnicamente relevantes. Efectos adicionales, observados en otros estudios, incluyeron prolongación del intervalo QTc, disminución de la FEVI, hipertrofia pituitaria y atrofia testicular tubular, matriz mesangial aumentada en el rińón, hemorragia en el tracto GI y la mucosa oral e hipertrofia de las células pituitarias anteriores. Se piensa que los cambios en el śtero (atrofia endometrial) y en la placa de crecimiento del hueso (engrosamiento epifisario o displasia del cartķlago), estarķan relacionados con la acción farmacológica del sunitinib. La mayorķa de estos hallazgos fueron reversibles, después de 2 a 6 semanas sin tratamiento. Genotoxicidad: El potencial genotóxico del sunitinib fue evaluado in vitro e in vivo. Sunitinib no fue mutagénico en bacterias, usando la activación metabólica brindada por el hķgado de rata. Sunitinib no indujo aberraciones cromosómicas estructurales en células linfocķticas de sangre periférica humana in vitro. En linfocitos de sangre periférica humana in vitro se observó poliploidia (aberraciones cromosómicas numéricas), tanto en presencia como en ausencia de activación metabólica. Sunitinib no fue clastogénico en la médula ósea de la rata in vivo. El metabolito activo principal no fue evaluado para determinar su potencial genotóxico. Carcinogenicidad: En 1 mes, el estudio de bśsqueda de rango de dosis por alimentación por sonda oral (0, 10, 25, 75 o 200 mg / kg / dķa) con dosis diarias continua en ratones transgénicos rasH2, el carcinoma y la hiperplasia de Brunner en las glįndulas del duodeno se han observados en pruebas en dosis mįs altas (200 mg / kg / dķa). A 6 meses, el estudio de carcinogenicidad por alimentación por sonda oral (0, 8, 25, 75 [reducido a 50] mg / kg / dķa), con una dosificación diaria se llevó a cabo en ratones transgénicos rasH2. Se observaron carcinomas gastroduodenales, un aumento en la incidencia de hemangiosarcomas de fondo, y / o hiperplasia de la mucosa gįstrica a dosis de ≥25 mg / kg / dķa después de 1 - o 6 meses de duración (≥ 7.3 veces el AUC en pacientes que recibieron la RDD). La relevancia de estos hallazgos de la carcinogenicidad observados en ratones trasgénicos rasH2 para humanos siguiendo un tratamiento con sunitinib de 1 a 6 meses no es clara. Toxicidad reproductiva y del desarrollo: No se observaron efectos sobre la fertilidad en machos o hembras en los estudios de toxicidad reproductiva. Sin embargo, en los estudios de toxicidad de dosis repetidas realizados en ratas y monos, se observaron efectos sobre la fertilidad femenina bajo la forma de atresia folicular, degeneración del cuerpo lśteo, cambios endometriales en el śtero y disminución del peso del śtero y ovarios, con niveles de exposición sistémica clķnicamente relevantes. Los efectos sobre la fertilidad masculina se vieron bajo la forma de atrofia tubular en los testķculos, reducción de los espermatozoides en los epidķdimos y depleción del coloide en la próstata y vesķculas seminales, con niveles de exposición plasmįtica 18 veces mįs altos que los observados en la clķnica. En las ratas, la mortalidad embriofetal relacionada con el tratamiento fue evidente en términos de reducciones significativas en el nśmero de fetos vivos, el mayor nśmero de resorciones (tempranas y totales), el incremento correspondiente en la pérdida post-implantación y la pérdida de la camada completa en 8 de 28 hembras preńadas, con niveles de exposición plasmįtica 5.5 veces mįs altos que los observados en la clķnica. En los conejos, las disminuciones de los pesos de los śteros grįvidos y del nśmero de fetos vivos, se debieron al aumento del nśmero de las resorciones, al incremento en la pérdida post-implantación y a la pérdida de la camada completa en 4 de 6 hembras preńadas, con niveles de exposición plasmįtica 3 veces mįs altos que los observados en la clķnica. El tratamiento con sunitinib en ratas durante la organogénesis, resultó en efectos sobre el desarrollo a 5 mg/kg/dķa, consistente de incidencia aumentada de malformaciones esqueléticas fetales, caracterizadas predominantemente como osificación retardada de las vértebras torįcicas/lumbares y ocurrieron con niveles de exposición plasmįtica 6 veces mįs altos que los observados en la clķnica. En los conejos, los efectos sobre el desarrollo consistieron en una mayor incidencia de labio hendido, con niveles de exposición plasmįtica aproximadamente iguales a los observados en la clķnica y labio hendido y paladar hendido, con niveles de exposición plasmįtica 2.7 veces mįs altos que los observados en la clķnica. No se efectuó un estudio definitivo de toxicidad en el desarrollo embriofetal en conejos, ya que los efectos embriofetales quedaron demostrados claramente en la rata y se reportaron en el estudio preliminar efectuado en conejos.
Interacciones Medicamentosas: Estudios de interacción se han realizado sólo en adultos. Drogas que pueden aumentar las concentraciones plasmįticas del sunitinib: La administración concomitante de sunitinib con el potente inhibidor de la CYP3A4 ketoconazol, resultó en un incremento de 49% y de 51% de los valores de Cmax y AUC0- del complejo [sunitinib + metabolito activo principal], respectivamente, después de la administración de una dosis śnica de sunitinib a voluntarios sanos. La administración de sunitinib con inhibidores potentes de la familia CYP3A4 (por ej. ritonavir, itraconazol, eritromicina, claritromicina, jugo de pomelo) puede aumentar las concentraciones de sunitinib. Por lo tanto, se deberķa evitar la administración concomitante con estos inhibidores o se debe considerar la selección de una medicación concomitante alterna que no inhiba, o tenga un potencial mķnimo para inhibir, a la CYP3A4. Si esto no es posible, puede ser necesaria la reducción de la dosis de SUTENT al mķnimo de 37.5 mg diarios para GIST y CMCR o 25 mg diarios para pNET, basados en el cuidadoso monitoreo de la tolerabilidad. Drogas que pueden disminuir las concentraciones plasmįticas de sunitinib: La administración concomitante de sunitinib con el inductor de la CYP3A4 rifampicina, resultó en una disminución de 23% y 46% de los valores de Cmax y AUC0-∞ del complejo [sunitinib + metabolito activo principal], respectivamente, después de la administración de una dosis śnica de sunitinib a voluntarios sanos. La administración del sunitinib con inductores potentes de la familia CYP3A4 (por ej., dexametasona, fenitoķna, carbamazepina, rifampicina, fenobarbital o Hypericum perforatum, también conocido como hierba de San Juan), puede disminuir las concentraciones de sunitinib. Por consiguiente, se debe evitar la administración concomitante con inductores, o se debe considerar la selección de una medicación concomitante alterna que no induzca, o tenga un potencial mķnimo para inducir a la CYP3A4. Si esto no es posible, puede ser necesario un aumento de la dosis de Sutent en 12.5 mg (hasta 87.5 mg al dķa para GIST y CMCR o 62.5 mg al dķa para pNet), basado en un monitoreo cuidadoso de la tolerabilidad.
Sobredosificación: No existe ningśn antķdoto especķfico para la sobredosificación con sunitinib y el tratamiento de las sobredosis deberķa consistir en las medidas de soporte generales. Si estį indicado, se puede intentar la eliminación de la droga no absorbida por emesis o lavado gįstrico. Se han reportado algunos casos de sobredosis; éstos fueron asociados con o sin reacciones adversas consistentes con el perfil de seguridad de sunitinib. Peso, calidad de vida: Anįlisis de datos demogrįficos farmacocinéticos de la población indicaron que son necesarios ajustes de la dosis inicial para el peso o para la calidad de vida mediante Eastern Cooperative Oncology Group (ECOG). Género: La información disponible indicó que las mujeres podrķan tener cerca de un 30% menos de depuración aparente (CL/F) de sunitinib que los hombres: esta diferencia, sin embargo, no necesita ajuste inicial de dosis.
Observaciones:Fertilidad, embarazo y lactancia:Embarazo: No se han realizado estudios en mujeres embarazadas usando Sutent. Los estudios en animales han evidenciado toxicidad reproductiva, incluyendo malformaciones fetales. Sutent no deberķa usarse en el embarazo, ni en ninguna mujer que no esté empleando contracepción adecuada, a menos que el posible beneficio justifique el riesgo potencial para el feto. Si se usa Sutent durante el embarazo, o si la paciente queda embarazada mientras estį recibiendo Sutent, la paciente debe ser informada de los posibles riesgos para el feto. A las mujeres en capacidad de procrear (en edad fértil), se les debe aconsejar un método anticonceptivo efectivo y evitar quedar embarazadas, mientras estén recibiendo tratamiento con Sutent. Sutent (0.3; 1.0; 3.0 mg/kg/dķa) se evaluó en un estudio de desarrollo pre y post-natal en ratas embarazadas. Se redujo la ganancia de peso en el cuerpo materno durante la gestación y lactancia en >1 mg/kg/dķa, pero no se observó toxicidad reproductiva materna en hasta 3 mg/kg/dķa (exposición estimada >2.3 veces la AUC en pacientes que recibieron la dosis diaria recomendada (DDR). Se observaron reducciones en los pesos corporales de las crķas durante los perķodos de pre y post-destete a 3 mg/kg/dķa. No se observó toxicidad en el desarrollo a 1 mg/kg/dķa (exposición aproximada =0.9 veces el AUC en pacientes que recibieron la DDR). Lactancia: Sunitinib y/o sus metabolitos se excretan en la leche de ratas. No se sabe si sunitinib o su metabolito activo principal se excretan en la leche humana. Como las drogas se excretan comśnmente en la leche humana y considerando el potencial de reacciones adversas serias en los infantes lactantes, las mujeres no deben amamantar mientras estén tomando Sutent. Fertilidad: De acuerdo con hallazgos no clķnicos, la fertilidad masculina y femenina podrķa verse comprometida por el tratamiento con Sutent.
 Laboratorio
Laboratorio