Composición: Cada comprimido recubierto contiene: Talidomida 100 mg. Excipientes: Celulosa Microcristalina, Lactosa Monohidrato, Dióxido de Silicio Coloidal, Povidona, Croscarmelosa Sódica, Estearato de Magnesio, Hipromelosa, Macrogol 400, Polisorbato 80, Dióxido de Titanio.
Acción Terapéutica: Otros inmunosupresores.
Indicaciones:Mieloma múltiple (MM): Talidomida en combinación con dexametasona está indicada como tratamiento de pacientes con mieloma múltiple recientemente diagnosticado. Eritema nodoso leproso (ENL): Talidomida está indicada para el tratamiento agudo de las manifestaciones cutáneas del ENL moderadas o severas. Talidomida no está indicada como monoterapia para el tratamiento de ENL en presencia de neuritis moderada a severa. Talidomida también está indicada como terapia de mantenimiento para la prevención y supresión de las manifestaciones cutáneas recurrentes de ENL.
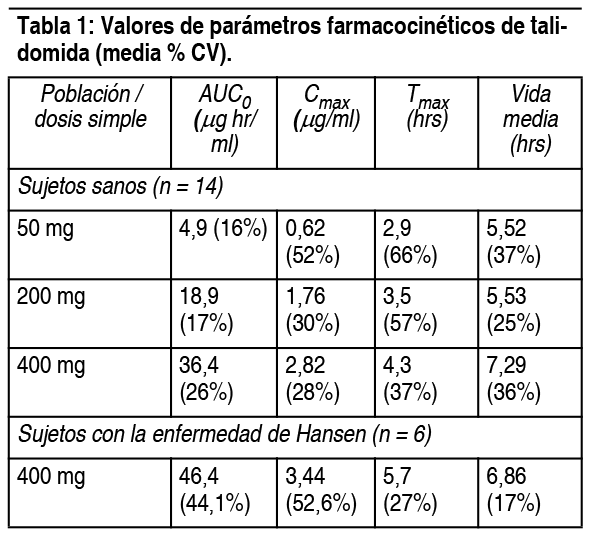
Propiedades:Farmacología: Mecanismo de acción: El mecanismo de acción de la talidomida no es completamente entendido. La talidomida tiene propiedades inmunomoduladoras, antiinflamatorias y antiangiogénicas. Los datos disponibles de estudios in vitro y ensayos clínicos sugieren que los efectos inmunológicos de este compuesto pueden variar sustancialmente bajo diferentes condiciones, pero pueden estar relacionados con la supresión de la producción excesiva del factor de necrosis tumoral alfa (TNF-α) y la modulación de moléculas de adhesión celular implicadas en la migración de los leucocitos. Por ejemplo, la administración de la talidomida reduciría los niveles circulantes de TNF-α en pacientes con eritema nudoso leproso (ENL), aunque también se ha demostrado que aumentaría los niveles en plasma de TNF-α en pacientes VIH-seropositivos. Otras propiedades antiinflamatorias e inmunomoduladoras de la talidomida pueden incluir la supresión de la participación de los macrófagos en la síntesis de prostaglandinas, y la modulación de la IL-10 y la IL-12 por células mononucleares de sangre periférica. El tratamiento con talidomida de los pacientes con mieloma múltiple es acompańado por un aumento en el número de las células natural killer en la circulación, y un aumento en los niveles plasmáticos de IL-2 e interferón gamma (citoquinas derivadas de células T asociadas con actividad citotóxica). La talidomida inhibió la angiogénesis en un modelo de arteria umbilical humana en estudios in vitro. Los procesos celulares de angiogénesis inhibida por la talidomida pueden incluir la proliferación de células endoteliales. Destino en el organismo (farmacocinética): Absorción: La biodisponibilidad absoluta de talidomida a partir de las cápsulas no ha sido caracterizada aún en humanos debido a su escasa solubilidad en agua. En estudios realizados tanto en voluntarios sanos como enfermos con enfermedad de Hansen, el tiempo medio hasta las concentraciones plasmáticas pico (Tmax) de talidomida osciló entre 2.9 a 5.7 horas, indicando que talidomida es absorbida lentamente del tracto gastrointestinal. Cómo la extensión de la absorción [como es medida por el área bajo la curva (AUC)] es proporcional a la dosis en sujetos sanos, el pico de concentración (Cmax) observado aumentó en forma menos que proporcional (ver Tabla 1). Esta ausencia de proporcionalidad de la dosis Cmax, combinada con el incremento observado de los valores Tmax, sugiere que la escasa solucbilidad de talidomida en medio acuoso puede estar entorpeciendo la velocidad de absorción.
La coadministración de talidomida con una comida de alto contenido graso produce cambios menores (<10%) de la AUC y valores Cmax observados; produce un incremento de la Tmax en aproximadamente 6 horas. Distribución: No hay referencias sobre el porcentaje de combinación proteica de la talidomida en el plasma y se desconoce si esta presente en el líquido de eyaculación en el hombre. Metabolismo: No se conoce con certeza el destino metabólico de la talidomida en humanos. No aparece como metabolizada ampliamente por el hígado pero aparece como sometida a hidrólisis no-enzimática en plasma a múltiples metabolitos. En un estudio de dosis repetida en que se aplicó talidomida 200 mg a 10 mujeres sanas durante 18 días, talidomida desplegó perfiles farmacocinéticos similares el primero y el último día de dosificación. Esto sugiere que la talidomida no induce ni inhibe su propio metabolismo. Eliminación: Como se indica el cuadro 1, la vida media de eliminación promedio oscila entre 5 a 7 horas después de una única dosis y no es alterada después de dosis múltiples. Se desconoce por ahora el destino metabólico preciso y la ruta de eliminación de la talidomida en humanos. La talidomida en sí tiene una excreción renal de 1.15 ml/minuto con menos del 0.7% de la dosis excretada en orina como droga inalterada. Después de una única dosis, los niveles de talidomida en orina no fueron detectables a las 48 horas post-dosis. Si bien se cree que la talidomida es hidrolizada a una cantidad de metabolitos, solamente una pequeńa cantidad (0.02 % de la dosis administrada) de 4-OH-talidomida fue identificada en orina de los sujetos a las 12 a 24 horas después del dosaje. Farmacocinética en poblaciones especiales: Sujetos HIV-seropositivos: No hay una aparente diferencia significativa de los valores paramétricos farmacocinéticos medidos entre sujetos humanos sanos y sujetos HIV-seropositivos después de la administración de una única dosis de talidomida comprimidos recubiertos. Pacientes con la enfermedad de Hansen: Los pacientes con la enfermedad de Hansen en referencia con sujetos sanos, pueden presentar una biodisponibilidad aumentada de talidomida. Este incremento es reflejado tanto en un aumento del área bajo la curva y niveles pico plasmáticos aumentados. Se desconoce el significado clínico de este incremento. Pacientes con insuficiencia renal: La farmacocinética de talidomida en pacientes con insuficiencia renal no ha sido determinada. En un estudio de 6 pacientes con enfermedad renal en etapa terminal, talidomida (200 mg/día) fue administrada en un día de no diálisis y en uno de diálisis, se compararon los perfiles de concentración-tiempo en un día de no diálisis y durante la diálisis. Las muestras de sangre tomadas al menos 10 horas después de la dosis, mostraron que el aclaramiento total promedio aumentó en un factor de 2.5 durante la hemodiálisis. Debido a que la diálisis se realizó 10 horas después de la administración de la dosis, las curvas de concentración tiempo de la droga no mostraron diferencias estadísticamente significativas para los días que los pacientes estaban dentro y fuera de la diálisis. Por lo tanto, no se requiere ajustar dosis en pacientes con insuficiencia renal dializados. Pacientes con enfermedad hepática: No se ha determinado la farmacocinética de talidomida en pacientes con dańo hepático. Edad: El análisis de la información en base a estudios farmacocinéticos en voluntarios sanos y pacientes con enfermedad de Hansen, con edades entre 20 y 69 ańos. Sexo: Mientras no se ha realizado un estudio comparativo sobre los efectos de la farmacocinética de la talidomida, el examen de la información sobre talidomida no revela diferencias significativas por sexo en los valores farmacocinéticos.
Posología: La prescripción de medicamentos a las mujeres en edad fértil debe estar supeditada a la confirmación inicial y continua de resultados de las pruebas de embarazo negativos. Se debe considerar la posibilidad de reducir la dosis, retrasar o interrumpir el tratamiento en los pacientes que desarrollan reacciones adversas según los criterios de toxicidad común del Instituto Nacional del Cáncer (CTC INC) de grado 3 ó 4 y/o según el criterio clínico. Mieloma múltiple: Talidomida se administra en combinación con dexametasona en ciclos de tratamiento de 28 días. La dosis de talidomida es de 200 mg por vía oral 1 vez al día con agua, preferiblemente al acostarse y después de al menos 1 hora de la cena. La dosis de dexametasona es de 40 mg al día por vía oral en los días 1-4, 9-12, y 17-20, cada 28 días. Los pacientes que desarrollan efectos secundarios como constipación, somnolencia o neuropatía periférica pueden beneficiarse de forma temporal si discontinúan el medicamento o lo continúan con una dosis menor. Con la reducción de estos efectos secundarios, se puede recomenzar a tomar la droga con una dosis más baja o con la dosis anterior según el criterio clínico. Eritema nudoso leproso: Ante un episodio de ENL cutánea, la dosificación con talidomida debe iniciarse con dosis de 100 a 300 mg/día, administrada 1 vez al día, con agua, preferiblemente al acostarse y al menos 1 hora después de la cena. Los pacientes que pesen menos de 50 kg deben comenzar con el extremo inferior del intervalo de dosis. En los pacientes con una reacción cutánea severa de ENL, o en aquellos que han requerido mayores dosis para controlar la reacción, la dosificación de talidomida se puede iniciar con dosis más altas de hasta 400 mg/día 1 vez al día al acostarse o en dosis divididas, con agua y por lo menos 1 hora después de las comidas. En pacientes con neuritis moderada a severa asociada a una reacción de ENL grave, los corticosteroides pueden iniciarse de forma concomitante con talidomida. El uso de esteroides se puede reducir y discontinuar cuando la neuritis mejora. La dosificación con talidomida por lo general se debe continuar hasta que los signos y síntomas de reacción activa han disminuido, por lo general en un período de al menos 2 semanas. Luego, los pacientes pueden disminuir la dosis de la medicación de a 50 mg cada 2 a 4 semanas. Los pacientes que tienen una historia documentada de requerir tratamiento de mantenimiento prolongado para evitar la recurrencia de ENL cutánea o que tuvieron recaídas, deben mantener la dosis mínima necesaria para controlar la reacción. La disminución del medicamento debe intentarse cada 3 a 6 meses, en decrementos de 50 mg cada 2 a 4 semanas.
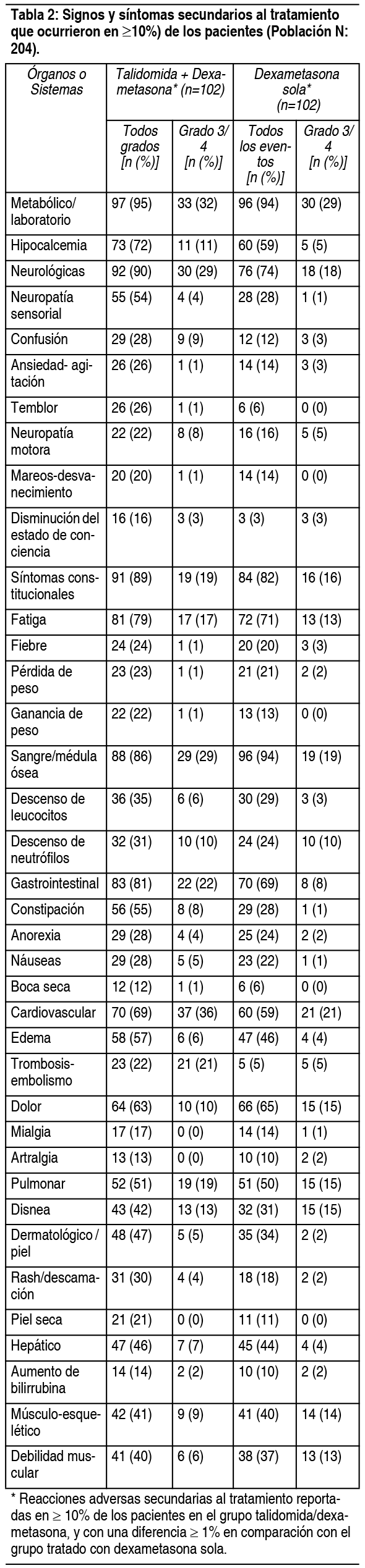
Efectos Colaterales: La reacción adversa más seria asociada con la talidomida es su teratogenicidad humana documentada (ver Contraindicaciones). El riesgo de severos defectos congénitos, básicamente facomielia o muerte del feto, es extremadamente alto durante el período crítico del embarazo. El período crítico es estimado, dependiendo de la información, entre los 35 y los 50 días después del último período menstrual. El riesgo de defectos congénitos potencialmente severos fuera de este período crítico es desconocido, pero puede ser importante. Basado en este conocimiento, talidomida no debe ser usada en ningún momento durante el embarazo. Dado que la talidomida está presente en el semen de los pacientes que recibieron la droga, los hombres que fueron tratados con talidomida deben utilizar siempre preservativos de látex durante las relaciones sexuales con mujeres en edad fértil, inclusive si se les ha practicado una vasectomía exitosa. La talidomida se ha estudiado en ensayos clínicos controlados y no controlados en pacientes con mieloma múltiple y ENL y en las personas seropositivas al VIH. Además, la talidomida se ha administrado investigacionalmente por más de 20 ańos en numerosas indicaciones. Los perfiles de eventos adversos de estos usos se resumen más adelante. Eventos adversos en un ensayo clínico controlado con mieloma múltiple: El análisis de seguridad se llevó a cabo en 204 pacientes que recibieron el fármaco en estudio clínico aleatorizado. La Tabla 2 enumera los signos y síntomas secundarios al tratamiento que ocurrieron en ≥10% de los pacientes. Los acontecimientos adversos más frecuentes fueron fatiga, hipocalcemia, edema, constipación, neuropatía sensorial, disnea, debilidad muscular, leucopenia, neutropenia, rash/descamación, confusión, anorexia, náuseas, ansiedad/agitación, temblores, fiebre, pérdida de peso, trombosis/embolia, neuropatía motora, ganancia de peso, mareo y piel seca. Veintitrés por ciento de los pacientes (47/204) interrumpieron el tratamiento debido a eventos adversos, el 30% (31/102) de los tratados con talidomida y dexametasona y 16% (16/102) de los tratados con dexametasona sola.
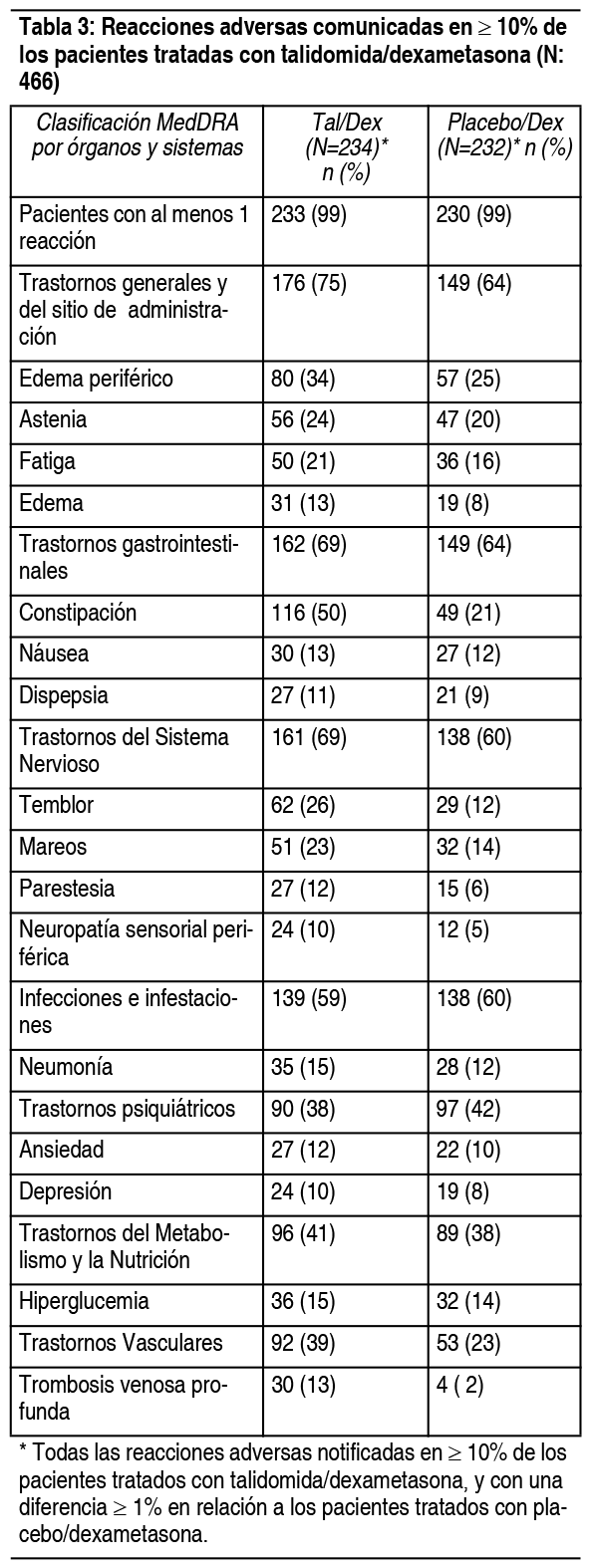
Otro análisis de seguridad se llevó a cabo en un estudio con 466 pacientes que recibieron tratamiento. La Tabla 3 enumera las reacciones adversas más comunes (≥10%) que se observaron. El cuadro 3 enumera las reacciones adversas más comunes de grado 3/4 (que ocurrieron en >2% de los pacientes). Las reacciones adversas más frecuentemente reportadas por los pacientes tratados con talidomida/dexametasona fueron constipación, edema periférico, temblor, astenia, mareos y fatiga. Las reacciones adversas con una frecuencia de por lo menos 2 veces mayores en el grupo de talidomida dexametasona que en el grupo placebo/dexametasona incluyen constipación, temblor, trombosis venosa profunda y neuropatía sensorial periférica. El 26% de los pacientes (121/466) debió suspender al tratamiento a causa de eventos adversos; 37% (86/234) de los tratados con talidomida/dexametasona y 15% (35/232) de los tratados con placebo/dexametasona.
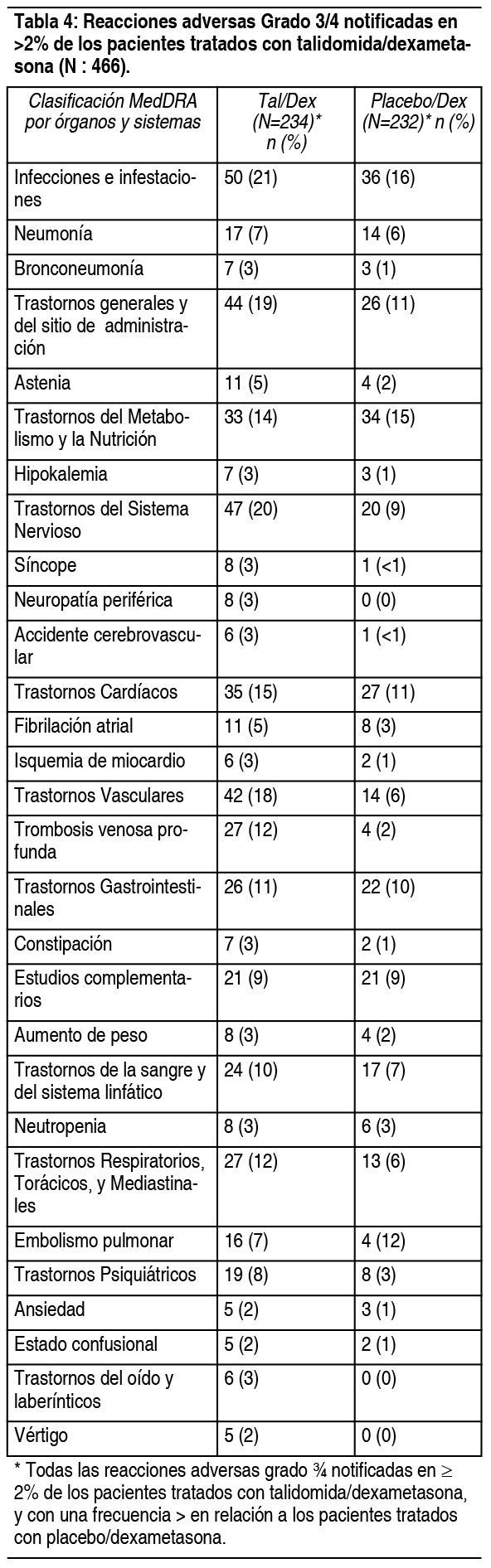
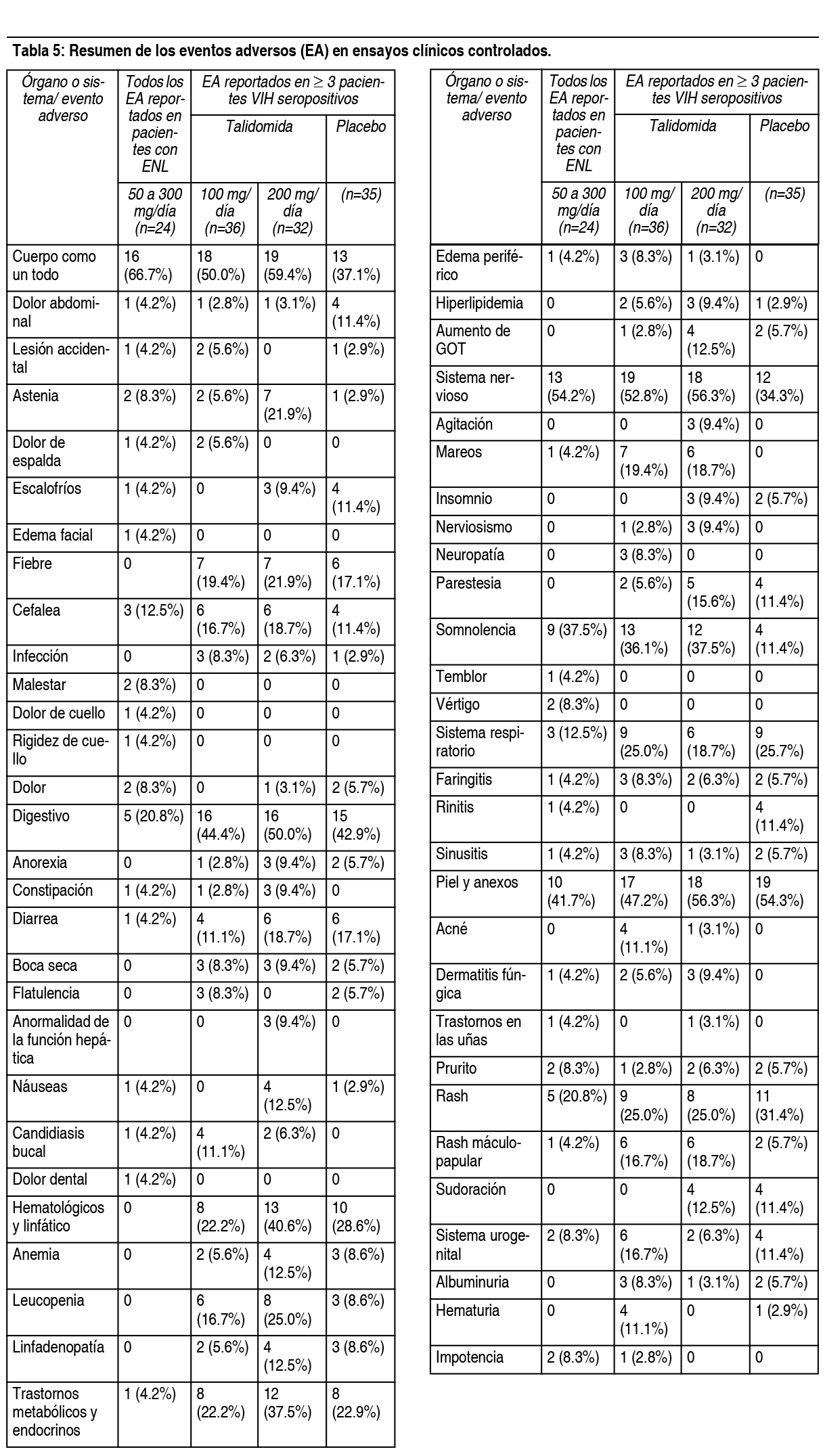
Reacciones adversas menos comunes en estudios clínicos controlados en pacientes con mieloma múltiple: Las siguientes reacciones adversas no fueron descritas anteriormente*: Trastornos gastrointestinales: Vómitos, sequedad de boca, peritonitis, perforación diverticular. Trastornos del sistema nervioso: Somnolencia, hipoestesia, polineuropatía, ataque isquémico transitorio. Trastornos respiratorios, torácicos y mediastínicos: Bronquitis. Trastornos psiquiátricos: Alteración del estado de ánimo. Trastornos vasculares: Hipotensión, hipotensión ortostática. Trastornos cardíacos: Bradicardia. Trastornos oculares: Visión borrosa. * Todas las reacciones adversas que ocurrieron en ≥3 % de los pacientes tratados con talidomida/dexametasona, y con una diferencia ≥1 % en comparación con el grupo tratado con placebo/dexametasona. Todas las reacciones adversas de grado 3/4 y graves notificadas en > 2 pacientes tratados con talidomida/dexametasona, y con un porcentaje mayor en comparación con el grupo tratado con placebo/dexametasona se han considerado para su posible inclusión. En todos los casos se ha aplicado el criterio médico para la consideración de la evaluación de la causalidad. Incidencia en ensayos clínicos controlados en pacientes con ENL: La Tabla 5 enumera los signos y los síntomas secundarios al tratamiento que ocurrieron en pacientes con ELN tratados con talidomida en ensayos clínicos controlados. Las reacciones adversas más comunes (≥10%) reportados en pacientes con ENL fueron: somnolencia, erupción cutánea, dolor de cabeza. Las dosis oscilaron entre 50 a 300 mg/día. Todos los eventos adversos fueron de intensidad leve a moderada, y ninguno requirió suspender el tratamiento
Otros efectos adversos observados en pacientes con ENL: La talidomida en dosis de hasta 400 mg/día fue administrada en los Estados Unidos durante un período de 19 ańos en 1465 pacientes con ENL. La literatura publicada describe el tratamiento de un número adicional de 1.678 pacientes. Para proveer una estimación significativa de la proporción de personas que tienen reacciones adversas, los tipos de eventos similares se agruparon en un número menor de categorías estándar utilizando una versión modificada del diccionario/terminología COSTART. Estas categorías se utilizan en la lista a continuación. Todos los hechos fueron reportados, excepto aquellos que ya figuraban en la tabla anterior. Debido al hecho de que estos datos se obtuvieron de estudios no controlados, la tasa de incidencia no se puede determinar. Como se mencionó anteriormente, la relación causal entre la talidomida y estos eventos no pudo ser determinado en forma concluyente. Se trata de informes de todos los eventos adversos observados por los investigadores en los pacientes a los que habían administro la talidomida. Cuerpo en general: Abdomen distendido, fiebre, fotosensibilidad, dolor en las extremidades superiores. Sistema cardiovascular: Bradicardia, hipertensión, hipotensión, trastornos vasculares periféricos, taquicardia, vasodilatación. Sistema digestivo: Anorexia, aumento del apetito/aumento de peso, sequedad de boca, dispepsia, hepatomegalia, eructos, flatulencia, aumento de las enzimas hepáticas, obstrucción intestinal, vómitos. Hematológico y linfático: Disminución de velocidad de sedimentación globular, eosinofilia, granulocitopenia, anemia hipocrómica, leucemia, leucocitosis, leucopenia, VCM elevado, RBC anormal, bazo palpable, trombocitopenia. Metabólicos y endocrinos: Secreción inapropiada de ADH, amiloidosis, bilirrubinemia aumentada, incremento del nitrógeno ureico, creatinina aumentada, cianosis, diabetes, edema, alteraciones electrolíticas, hiperglucemia, hiperpotasemia, hiperuricemia, hipocalcemia, hipoproteinemia, aumento de LDH, hipofosfatemia, aumento de GPT. Músculo-esquelético: Artritis, dolor óseo, hipertonía, alteraciones articulares, calambres en las piernas, mialgia, miastenia, trastornos del periostio. Sistema nervioso: Alteraciones del pensamiento, agitación, amnesia, ansiedad, causalgia, parestesia peribucal, confusión, depresión, euforia, hiperestesia, insomnio, nerviosismo, neuralgia, neuritis, neuropatía, parestesia, neuritis periférica, psicosis. Sistema respiratorio: Tos, enfisema, epistaxis, embolia pulmonar, estertores, infección respiratoria, alteraciones de la voz. Piel y anexos: Acné, alopecia, piel seca, eczema, dermatitis exfoliativa, ictiosis, engrosamiento perifolicular, necrosis cutánea, seborrea, sudoración, urticaria, erupción vesicular. Sentidos especiales: Ambliopía, sordera, sequedad ocular, dolor ocular, tinnitus. Urogenital: Disminución del aclaramiento de creatinina, hematuria, orquitis, proteinuria, piuria, aumento de la frecuencia urinaria. Otros efectos adversos observados en pacientes VIH seropositivos: Además de los ensayos clínicos controlados, talidomida se ha utilizado en estudios no controlados con 145 pacientes. En estos pacientes VIH seropositivos tratados con talidomida se han reportado menos eventos adversos y se agruparon en un número menor de categorías estandarizadas usando el diccionario/terminología modificada COSTART y estas categorías son utilizadas en la lista a continuación. Los eventos adversos que se han incluido anteriormente en las otras tablas, o los que son demasiado generales para ser de carácter informativo, no están en la lista. Cuerpo en general: Ascitis, SIDA, reacción alérgica, celulitis, dolor torácico, escalofríos y fiebre, quistes, disminución del recuento de CD4, edema facial, gripe, hernia, alteración de los niveles de hormona tiroidea, moniliasis, reacción de fotosensibilidad, sarcoma, sepsis, infección viral. Sistema cardiovascular: Angina de pecho, arritmia, fibrilación auricular, bradicardia, isquemia cerebral, accidente cerebrovascular, insuficiencia cardíaca congestiva, tromboflebitis profunda, paro cardíaco, insuficiencia cardíaca, hipertensión, hipotensión, soplos, infarto de miocardio, palpitaciones, pericarditis, trastorno vascular periférico, hipotensión postural, síncope, taquicardia, tromboflebitis y trombosis. Sistema digestivo: Colangitis, ictericia colestásica, colitis, dispepsia, disfagia, esofagitis, gastroenteritis, trastornos gastrointestinales, hemorragia gastrointestinal, trastornos de las encías, hepatitis, pancreatitis, parotiditis, periodontitis, estomatitis, decoloración de la lengua, trastornos dentales. Hematológico y linfático: Anemia aplásica, anemia macrocítica, anemia megaloblástica, anemia microcítica. Metabólicos y endocrinos: Avitaminosis, bilirrubinemia, deshidratación, hipoglucemia hipercolesterolemia, aumento de fosfatasa alcalina, aumento de lipasa, aumento de creatinina sérica, edema periférico. Músculo-esquelético: Mialgia, miastenia. Sistema nervioso: Marcha anormal, ataxia, disminución de la libido, disminución de los reflejos, demencia, disestesia, discinesia, labilidad emocional, hostilidad, hipoalgesia, hipercinesia, falta de coordinación, meningitis, trastorno neurológico, temblor, vértigo. Sistema respiratorio: Apnea, bronquitis, enfermedad pulmonar, edema pulmonar, neumonía (incluyendo la neumonía por Pneumocystis carinii), rinitis. Piel y anexos: Angioedema, neoplasia benigna de la piel, eccemas, herpes simple, síndrome de Stevens-Johnson, trastornos de las uńas, psoriasis, prurito, decoloración de la piel, trastornos de la piel. Sentidos especiales: Conjuntivitis, trastornos oculares, trastornos de lagrimeo, retinitis, alteración del gusto. Otros efectos adversos observados en el uso post-comercialización: Sistema cardiovascular: Arritmias cardíacas, incluyendo fibrilación auricular, bradicardia, taquicardia, síndrome del seno enfermo, anormalidades en el ECG, infarto de miocardio. Sistema digestivo: Perforación intestinal, perforación gastrointestinal, obstrucción intestinal. Metabólicos y endocrinos: Desequilibrio en los electrolitos incluyendo hipercalcemia o hipocalcemia, hiperpotasemia e hipopotasemia, hiponatremia, hipotiroidismo, aumento de fosfatasa alcalina, síndrome de lisis tumoral. Sistema nervioso: Cambios en el estado mental o estado de ánimo como depresión e intentos de suicidio, trastornos de la consciencia incluyendo letargo, síncope, pérdida de la conciencia o estupor, convulsiones incluyendo tónico-clónica generalizadas y estado epiléptico, enfermedad de Parkinson. Piel y anexos: Eritema multiforme, necrólisis epidérmica tóxica. Hematológico y linfático: Disminución de glóbulos blancos, incluyendo neutropenia y neutropenia febril, cambios en el tiempo de protrombina, pancitopenia. Sistema respiratorio: Derrame pleural. Sistema reproductor y de la mama: Amenorrea, disfunción sexual. Trastornos del sistema inmunológico: Hipersensibilidad, angioedema/urticaria. Trastornos del oído y laberínticos: Deficiencia auditiva/sordera. Trastornos renales y urinarios: Insuficiencia renal. Otros eventos adversos descriptos en estudios publicados o reportados a partir de otras fuentes: Los siguientes eventos adversos han sido identificados, ya sea en la literatura publicada o en informes espontáneos de otras fuentes: insuficiencia renal aguda, amenorrea, estomatitis aftosa, obstrucción de los conductos biliares, túnel carpiano, leucemia mieloide crónica, diplopía, disestesias, disnea, enuresis, eritema nudoso, eritroleucemia, caída del pie, galactorrea, ginecomastia, efecto de resaca, hipomagnesemia, hipotiroidismo, linfedema, linfopenia, metrorragia, migrańa, mixedema, esclerosis nodular de la enfermedad de Hodgkin, nistagmo, oliguria, pancitopenia, petequias, púrpura, síndrome de Raynaud, úlcera de estómago, intento de suicidio, enfermedad pulmonar intersticial e infecciones graves (por ejemplo, sepsis fatal incluyendo shock séptico).
Contraindicaciones: Hipersensibilidad al principio activo o alguno de los constituyentes de la fórmula. Talidomida puede causar dańo fetal cuando se administra a una mujer embarazada. La talidomida está contraindicada en mujeres que están embarazadas. La talidomida es un potente teratogénico humano, que induce una alta frecuencia de defectos de nacimientos graves y potencialmente mortales, incluso después de una sola dosis. Se ha reportado mortalidad en, o poco después del nacimiento, en aproximadamente el 40% de los lactantes. Si se utiliza este medicamento durante el embarazo o si la paciente queda embarazada mientras está tomando este medicamento, la paciente debe ser informada del riesgo potencial para el feto. Si se produce un embarazo durante el tratamiento con talidomida, el fármaco debe suspenderse inmediatamente.
Advertencias:Cuidados especiales: Este medicamento no debe ser suministrado a mujeres embarazadas bajo ningún concepto. Tampoco debe administrarse a mujeres en edad fértil. Su ingesta produce malformaciones fetales. Toxicidad embriofetal: La talidomida es un potente teratógeno humano que induce una alta frecuencia de defectos de nacimientos graves y potencialmente mortales, incluso después de una sola dosis. Se ha reportado mortalidad en, o poco después del nacimiento, en aproximadamente el 40% de los lactantes. Cuando no existe un tratamiento alternativo satisfactorio, las mujeres con capacidad de reproducción pueden ser tratadas con talidomida siempre que tomen las precauciones adecuadas para evitar el embarazo. La ingestión oral es el único tipo de exposición materna a talidomida, conocida por causar defectos de nacimiento asociados con la droga. No hay datos específicos disponibles sobre los riesgos reproductivos de la absorción cutánea o inhalatoria de la talidomida. Sin embargo, las mujeres con capacidad de reproducción deben evitar el contacto con talidomida. Talidomida debe ser almacenado en envases tipo blister hasta su ingestión. Si hay contacto con las cápsulas de la talidomida no intactas o con el contenido en polvo, el área expuesta debe ser lavada con agua y jabón. Si el personal de salud u otros cuidadores se expusieran a fluidos corporales de los pacientes que recibieron talidomida, el área expuesta debe lavarse con agua y jabón. Deben ser utilizadas precauciones adecuadas, como el uso de guantes, para evitar la posible exposición cutánea a talidomida. Mujeres en edad fértil: Las mujeres con capacidad reproductiva deben evitar el embarazo durante al menos 4 semanas antes de comenzar la terapia con talidomida, durante la terapia, durante la interrupción de la dosis y durante al menos 4 semanas después de completar la terapia con talidomida. Las mujeres deben comprometerse, ya sea de abstenerse en forma continua a mantener relaciones sexuales heterosexuales, o de usar 2 métodos anticonceptivos confiables, una forma altamente efectiva de anticoncepción como ligadura de trompas, DIU, hormonales (píldoras anticonceptivas, inyecciones, parches hormonales, anillos vaginales o implantes) o la vasectomía de su pareja y un método anticonceptivo efectivo adicional (preservativo masculino de látex o un condón sintético, diafragma o capuchón cervical). La anticoncepción debe comenzar 4 semanas antes de iniciar el tratamiento con talidomida, y debe continuar durante la terapia, durante la interrupción de la dosis y durante 4 semanas después de la suspensión de la terapia con talidomida. La anticoncepción confiable está indicada incluso cuando ha habido historia de infertilidad, a menos que, se les haya practicado una histerectomía. Las mujeres con potencial reproductivo deben ser referidas a un profesional calificado en métodos anticonceptivos, de ser necesario. Las mujeres con potencial reproductivo deben tener 2 pruebas de embarazo negativas antes de iniciar el tratamiento con talidomida. La primera prueba se debe realizar de 10 a 14 días antes, y la segunda prueba dentro de las 24 horas antes de la prescripción de talidomida. Una vez que el tratamiento ha comenzado y durante las interrupciones de las dosis, se deben realizar pruebas de embarazo en las mujeres con potencial reproductivo, cada semana durante las primeras 4 semanas de uso, y luego se deben repetir cada 4 semanas en mujeres con ciclos menstruales regulares. Si los ciclos menstruales son irregulares, las pruebas de embarazo deben realizarse cada 2 semanas. Si la paciente pierde su período o si presenta alguna anormalidad en el sangrado menstrual se deben realizar pruebas de embarazo y dar consejo médico. El tratamiento con talidomida debe interrumpirse durante esta evaluación. Varones con capacidad reproductiva: La talidomida está presente en el semen de los pacientes que reciben el fármaco. Por lo tanto, los hombres siempre deben utilizar un preservativo de látex o sintético durante el contacto sexual con mujeres en edad fértil, mientras estén tomando talidomida y hasta por 28 días después de descontinuar el tratamiento, incluso si se han sometido a una vasectomía exitosa. Los pacientes varones que toman talidomida no deben donar esperma. Donación de sangre: Los pacientes no deben donar sangre durante el tratamiento con talidomida y durante 1 mes después de la discontinuación del mismo, debido a que la sangre puede ser transfundida a una paciente embarazada cuyo feto no debe ser expuesto a talidomida. Eventos trombóticos: Los pacientes tratados con talidomida presentan un mayor riesgo de tromboembolismo venoso (tales como trombosis venosa profunda y embolia pulmonar). Este riesgo aumenta significativamente cuando la talidomida se utiliza en combinación con agentes quimioterapéuticos estándares incluyendo dexametasona. En un ensayo controlado, la tasa de eventos tromboembólicos venosos fue 22.5% en los pacientes que recibieron talidomida en combinación con dexametasona, comparado con el 4.9% en pacientes tratados con dexametasona sola (p = 0.002). Los pacientes y los médicos deben estar atentos a los signos y síntomas de tromboembolismo. En un ensayo clínico en pacientes con MM no tratados previamente, en los que se administró talidomida y dexametasona vs. placebo y dexametasona también se reportó cardiopatía isquémica (11.1%), que incluía infarto de miocardio (1.3%), y accidente cerebrovascular (accidente cerebrovascular, 2.6%). Se debe considerar la profilaxis trombótica en base a una evaluación de los factores de riesgo subyacentes en los pacientes individuales. Los pacientes y los médicos deben estar atentos a los signos y síntomas de la enfermedad tromboembólica. Los pacientes deben ser instruidos para buscar atención médica si desarrollan síntomas tales como dificultad para respirar, dolor en el pecho, el brazo o la inflamación de la/s pierna/s. Se deben utilizar con precaución agentes que puedan aumentar el riesgo de tromboembolismo. Somnolencia y cansancio: La talidomida frecuentemente produce somnolencia y cansancio. Los pacientes deben ser instruidos a evitar situaciones en las que la somnolencia puede constituir un problema y de no asociar a otras medicaciones que puedan provocar somnolencia, sin el adecuado consejo médico Los pacientes deben ser advertidos en cuanto al posible deterioro de la capacidad mental y/o física necesaria para el desempeńo de las tareas peligrosas, como conducir un coche u operar maquinaria compleja o peligrosa. Puede ser necesaria una reducción de la dosis. Neuropatía periférica: La talidomida causa dańo nervioso que puede ser permanente y transformarse en un efecto colateral potencialmente severo que puede ser irrereversible. La neuropatía periférica ocurre generalmente después del uso crónico durante un período de meses; aunque también existen informes relacionados a su uso durante un período breve. La correlación con la dosis acumulada no es clara. Los síntomas pueden ocurrir a veces después de discontinuado el tratamiento con talidomida y pueden resolverse lentamente, o no resolverse de ninguna manera. Pocos informes de neuropatía han surgido durante el tratamiento de ENL, a pesar del prolongado período de tratamiento con talidomida. Sin embargo, la incapacidad clínica para diferenciar la neuropatía causada por la talidomida de la neuropatía producida en la enfermedad de Hansen dificulta determinar exactamente la incidencia de la neuropatía relacionada con talidomida en los pacientes con ENL tratados con talidomida. Los pacientes deben ser examinados a intervalos mensuales durante los 3 primeros meses de terapia con talidomida para permitirle al médico detectar los primeros signos de neuropatía, que incluyen entumecimiento, hormigueo o dolor de manos y pies. Luego los pacientes deben ser evaluados periódicamente y durante el tratamiento. Los pacientes deben ser examinados a intervalos mensuales durante los 3 primeros meses de terapia con talidomida para permitirle al médico detectar los primeros signos de neuropatía, que incluyen entumecimiento, hormigueo o dolor de manos y pies. Luego, los pacientes deben ser evaluados periódicamente durante el tratamiento. Los pacientes deben ser regularmente asesorados, interrogados y evaluados en busca de signos o síntomas periféricos de neuropatía. Se deben considerar las pruebas electrofisiológicas, que consisten en la medición de las amplitudes basales de los potenciales de acción nerviosos sensitivos (SNAP), y posteriormente bajo esfuerzo, cada 6 meses para detectar neuropatía asintomática. Si se desarrollaran síntomas de neuropatía inducida por talidomida se debe suspender inmediatamente su administración para limitar el dańo, si fuera clínicamente adecuado. Por lo general, el tratamiento con talidomida sólo debe reiniciarse si la neuropatía vuelve al estado basal. Los medicamentos que se conocen están asociados con neuropatía se deben utilizar con precaución en pacientes que reciben talidomida. Mareo e hipotensión ortostática: También se les debe informar a los pacientes que la talidomida puede causar mareo e hipotensión ortostática y que por ello deben sentarse durante unos pocos minutos antes de levantarse de una posición decúbito. Neutropenia: Puede producirse disminución del conteo de glóbulos blancos, incluyendo neutropenia, con la indicación de talidomida. El tratamiento no debe ser comenzado con un recuento de neutrófilos absolutos (RAN) de menos de 750/mm3. Los glóbulos blancos deben ser monitoreados cuali y cuantitativamente en forma permanente, especialmente en pacientes que pueden ser más propensos a la neutropenia, tales como los pacientes con serología positiva para HIV. Si el RAN disminuye a menos de 750/mm3 durante el tratamiento, el esquema terapéutico del paciente debe ser reevaluado y, si la neutropenia persiste, se debe considerar suspender el tratamiento con talidomida, si es clínicamente adecuado. Trombocitopenia: Se ha reportado trombocitopenia de grado 3 ó 4 en asociación con el uso clínico de la talidomida. Se deben realizar hemogramas, incluyendo recuento de plaquetas, para su control. Puede ser necesaria una reducción de la dosis, retrasar o interrumpir el tratamiento. Se deben vigilar los signos y síntomas de hemorragia, incluyendo petequias, epistaxis y hemorragia gastrointestinal, especialmente si la medicación concomitante puede aumentar el riesgo de sangrado. Carga viral-HIV aumentada: En un ensayo aleatorio, controlado a placebo de talidomida en una población de pacientes VIH- seropositivos, se observó que los niveles de VIH-RNA en plasma aumentaron (cambio medio = 0.42 log/10 copias VIH-RNA/ml, p = 0.04 comparado por placebo). Una tendencia similar fue observada en un segundo estudio no publicado realizado con pacientes que eran VIH-seropositivos. La importancia clínica de este aumento es desconocido. Ambos estudios se realizaron antes de la disponibilidad de la terapia antirretroviral de gran actividad. Hasta que la importancia clínica de este hallazgo sea entendido, en pacientes VIH-seropositivos, la carga viral debe medirse después del primer y tercer mes de tratamiento y posteriormente cada 3 meses.
Precauciones:Uso pediátrico: No se han establecido la seguridad y la eficacia en pacientes pediátricos menores de 12 ańos de edad. Uso geriátrico: Los pacientes > 65 que participaron en un estudio clínico con talidomida y dexametasona tuvieron una mayor incidencia de la fibrilación auricular, constipación, fatiga, náuseas, hipopotasemia, trombosis venosa profunda, hiperglucemia, embolia pulmonar, y astenia en comparación con pacientes más jóvenes. Insuficiencia renal: No se han realizado estudios clínicos con talidomida en pacientes con insuficiencia renal leve, moderada o grave. No se espera que la insuficiencia renal influya en la exposición al fármaco, ya que < 3.5 % de la dosis se excreta en la orina como fármaco inalterado. No es necesario ajustar la dosis en pacientes con insuficiencia renal o en pacientes en diálisis. Insuficiencia hepática: No se han realizado estudios clínicos en pacientes con insuficiencia hepática. Carcinogénesis, mutagénesis, teratogénesis y trastornos de la fertilidad: 2 ańos de estudios de carcinogenicidad fueron realizados en ratas y ratones. No se observaron efectos tumorigénicos relacionados con el compuesto en los niveles más altos de dosis de 3.000 mg/kg/día a ratones machos y hembras (38 veces mayor que la dosis diaria recomendada en humanos de 400 mg, basada en área de superficie corporal [ASC]), 3.000 mg/kg/día a ratas hembras (75 veces la dosis máxima en humanos, basada en ASC), y 300 mg/kg/día a ratas macho (7.5 veces la dosis máxima humana, basada en ASC). La talidomida no fue mutagénica ni genotóxica en los siguientes ensayos: mutación bacteriana inversa de Ames (S. typhimurium y E. coli), mutación de células de ovario de hámster chino (AS52/XPRT), y ensayo in vivo de micronúcleos de ratón. Los estudios de fertilidad se llevaron a cabo en conejos machos y hembras, sin efectos relacionados con el compuesto sobre los índices de apareamiento y la fertilidad en todos los niveles de dosis de talidomida oral, siendo la más alta de 100 mg/kg/día para hembras y 500 mg/kg/día para machos (aproximadamente 5 y 25 veces la dosis máxima en humanos, respectivamente, en base al ASC). Efectos patológicos e histopatológicos testiculares (clasificados como leves) se observaron en los conejos machos a dosis de 30 mg/kg/día (aproximadamente 1.5 veces la dosis máxima en humanos, basado en ASC). Embarazo: Talidomida puede causar dańo embriofetal cuando se administra a una mujer embarazada y está contraindicada durante el embarazo. Talidomida es un teratógeno humano, que induce una alta frecuencia de defectos graves y potencialmente mortales de nacimiento como: amelia (ausencia de extremidades), focomelia (extremidades cortas), hipoplasticidad ósea, ausencia de huesos, anomalías del oído externo (incluyendo anotia, micropinna, conductos auditivos externos pequeńos o ausentes), parálisis facial, anormalidades oculares (anoftalmos, microftalmia), y defectos congénitos del corazón. También se han documentado afectación del sistema digestivo y las vías urinarias, malformaciones genitales y mortalidad al momento o poco después del nacimiento, en aproximadamente el 40% de los lactantes. Incluso una sola dosis tomada por una mujer embarazada puede causar defectos de nacimiento. Si se utiliza este medicamento durante el embarazo o si la paciente se queda embarazada mientras está tomando este medicamento, la paciente debe ser informada del riesgo potencial para el feto. Si se produjera un embarazo durante el tratamiento, se debe suspender inmediatamente el medicamento. En estas condiciones, se debe remitir a la paciente a un obstetra/ginecólogo experimentado en toxicidad reproductiva para una evaluación y un asesoramiento. Un estudio de toxicidad reproductiva pre y post-natal se llevó a cabo en conejas embarazadas. La incidencia de abortos relacionados con el compuesto aumentó así como la fetotoxicidad con el nivel más bajo de la dosis oral de 30 mg/kg/día (aproximadamente 1.5 veces la dosis máxima en humanos, basada en ASC) y con todos los niveles de dosis más altas. La mortalidad neonatal en conejas lactantes fue elevada con dosis orales mayores de 150 mg/kg/día (aproximadamente 7.5 veces la dosis máxima en humanos, basada en ASC). No hubo retrasos en el desarrollo postnatal, incluyendo las funciones de aprendizaje y la memoria, con dosis por vía oral de 150 mg/kg/día a conejos lactantes hembras (las concentraciones medias de talidomida en la leche variaron desde 22 hasta 36 mg/ml). Lactancia: Se desconoce si la talidomida es excretada en la leche humana. Debido a que muchas drogas son excretadas en leche humana y debido al potencial de serias reacciones adversas en la lactancia que podrían ser producidas por la talidomida, se debe llegar a la decisión acerca de si se debe discontinuar la lactancia materna o discontinuar la droga, considerando la importancia de la droga para la madre.
Interacciones Medicamentosas: La talidomida no es un sustrato de las isoenzimas del citocromo P450 humanas (CYP450), por lo que no las inhibe ni induce in vitro. Por lo tanto, no se pueden anticipar las interacciones farmacocinéticas cuando talidomida se administra conjuntamente con medicamentos que son sustratos, inhibidores o inductores del citocromo P450. Opiáceos, antihistamínicos, antipsicóticos, ansiolíticos, y otros depresores del SNC (incluyendo el alcohol): Se debe evitar el uso de opiáceos, antihistamínicos, antipsicóticos, ansiolíticos, y otros depresores del SNC simultáneamente con talidomida, ya que pueden causar un efecto sedante aditivo. Fármacos bradicardizantes: El uso de fármacos que enlentecen la conducción cardíaca, concomitantemente con talidomida, puede causar un efecto bradicardizante aditivo por lo que debe utilizarse con precaución. Las drogas cardiovasculares que pueden causar bradicardia incluyen: antagonistas de los canales de calcio, betabloqueantes, bloqueantes adrenérgicos no selectivos y digoxina. Otros fármacos que pueden causar bradicardia incluyen: bloqueantes H2 (por ejemplo, famotidina, cimetidina), litio, antidepresivos tricíclicos y bloqueantes neuromusculares (succinilcolina). Una dosis única de digoxina no tiene ningún efecto sobre el perfil farmacocinético de la talidomida. La seguridad del uso concomitante a largo plazo de ambas drogas no ha sido evaluada. Fármacos que causan neuropatía periférica: El uso de fármacos que causan la neuropatía periférica (por ejemplo, bortezomib, amiodarona, cisplatino, docetaxel, paclitaxel, vincristina, disulfiram, fenitoína, metronidazol, alcohol) puede provocar un efecto aditivo y se debe utilizar con precaución. Anticonceptivos hormonales: Los anticonceptivos hormonales aumentan el riesgo de tromboembolismo. No se sabe si el uso concomitante de anticonceptivos hormonales aumenta aún más el riesgo de tromboembolismo con talidomida. La coadministración una dosis única de noretindrona y etinil estradiol con talidomida, no modificó la farmacocinética del anticonceptivo. Warfarina: El perfil farmacocinético y el cociente internacional normalizado (INR) para la warfarina, después de una dosis oral única de 25 mg, fueron similares con y sin la coadministración de talidomida 200 mg/día (en estado estacionario). La dosis única de warfarina no tuvo ningún efecto sobre el perfil farmacocinético de la talidomida. Fármacos que interfieren con los anticonceptivos hormonales: El uso concomitante de inhibidores de la proteasa del VIH, griseofulvina, modafinilo, penicilinas, rifampicina, rifabutina, fenitoína, carbamazepina, o ciertos suplementos a base de hierbas, tales como la hierba de San Juan con anticonceptivos hormonales, puede disminuir la eficacia de la anticoncepción hasta 1 mes después de suspender estas terapias concomitantes. Por lo tanto, las mujeres que requieren tratamiento con 1 o más de estos medicamentos deben usar 2 otros métodos eficaces o muy eficaces de anticoncepción mientras esté tomando talidomida. Tratamientos concomitantes que pueden aumentar el riesgo de tromboembolismo: Los agentes eritropoyéticos u otros agentes que pueden aumentar el riesgo de tromboembolismo, como las terapias que contienen estrógeno, se deben utilizar con precaución en pacientes con mieloma múltiple que reciben talidomida con dexametasona.
Sobredosificación:Síntomas y tratamiento de dosis excesivas: Hubo 3 casos de sobredosis informada, todos suicidios intentados. No hubo casos fatales informados en dosis de hasta 14.4 g; todos los pacientes se recuperaron sin secuelas informadas. No existe un antídoto específico para una sobredosis con talidomida. En caso de una sobredosis, los signos vitales del paciente deben ser monitoreados y deben realizarse cuidados de soporte apropiados para mantener la presión arterial y el estado respiratorio.
Presentaciones: Envases conteniendo 30 y 100 comprimidos.
 Laboratorio
Laboratorio