Composición: Cada cápsula contiene: Tamsulosina Clorhidrato 0.4 mg. Excipientes: Povidona, Sacarosa, Copolímero de Acido Metacrílico (Eudragit L100), Etilcelulosa, Almidón de Maíz, Triglicéridos de Cadena Media (Miglyol), Talco, Alcohol Metílico, Alcohol Isopropílico, Agua Purificada, Gelatina, Dióxido de Titanio, Colorante FD&C Azul N° 2, Oxido de Hierro Amarillo.
Acción Terapéutica: Bloqueante de los receptores alfa1.
Indicaciones: Tratamiento de los síntomas funcionales de la hiperplasia prostática benigna (HPB).
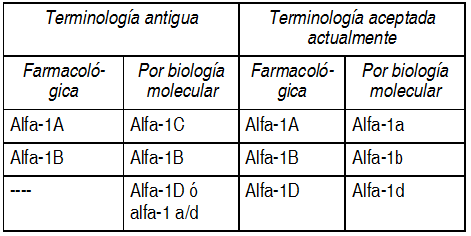
Propiedades:Acciones (farmacológica y/o terapéutica a los modos de acción del medicamento en el hombre): Los síntomas de HPB son causados por una obstrucción de la salida vesical (OSV) que, a su vez, puede estar causada por: 1. Una próstata agrandada. Esto comprime la uretra, incrementa la resistencia uretral y, de este modo, deteriora el flujo de salida urinario desde la vejiga. Esto se conoce como el componente mecánico o estático de la OSV. 2. Un incremento en el tono del músculo liso en el cuello de la vejiga, uretra prostática, cápsula prostática y adenoma (estroma) prostático debido a un incremento en la actividad del sistema nervioso simpático. Esto también aumenta la resitencia uretral y deteriora el flujo de salida urinario desde la vejiga. Este es el componente dinámico de la OSV. La obstrucción de salida vesical en particular, explica los síntomas obstructivos o de evacuación asociados con la HPB (por ej.; disminución en el tamańo y fuerza del chorro urinario, intermitencia y dificultad para el comienzo de la micción). Sin embargo, la OSV también puede provocar una inestabilidad secundaria del detrusor, que se cree que es la responsable de los síntomas irritativos o de llenado que se asocian a la HPB (por ej.; frecuencia, nocturna y urgencia). Los alfa-bloqueantes antagonizan el efecto de la noradrenalina (el neurotransmisor del sistema nervioso simpático) en los receptores alfa 1-adrenérgicos del cuello de la vejiga, uretra prostática, cáp`sula prostática y estroma prostático. Por consiguiente, reducen el componente dinámico de la OSV. Esto, junto con un posible efecto directo sobre la vejiga, son los responsables de la mejora en el flujo urinario y el alivio de los síntomas de HPB obtenidos con los alfa-bloqueantes. *Actividad antagonista en el receptoralfa-adrenérgico: Estudios previos que utilizaron al alfa-bloqueante fenoxibenzamina demostraron su eficacia en el tratamiento de los síntomas de la HPB. Sin embargo, la fenozibenzamina bloquea los receptores adrenérgicos alfa-1 y alfa-2 post-sinápticos y alfa-2 pre-sinápticos. Esta acción produce reacciones adversas en muchos pacientes, principalmente por causa del bloqueo de los receptores alfa2-adrenérgicos. Aunque el desarrollo de bloqueantes alfa1-selectivos como prazosina, terazosina, doxazosina y alfuzosina constituye un progreso, estos son solo selectivos para los receptores adrenérgicos alfa-1 sobre los receptores adrenérgicos alfa-2 y por lo tanto, tienen efectos en el sistema cardiovascular. El efecto del bloqueo de los receptores alfa1-adrenérgicos en el sistema cardiovascular puede causar vasodilatación de los vasos sanguíneos, que puede disminuir la presión arterial produciendo efectos relacionados no deseados (por ej., hipotensión postural y síncope). Desde fines de la década de los 80 ha quedado claro que existen varios subtipos de receptores alfa1-adrenérgicos. Hasta hace muy poco quedaba todavía una cierta confusión respecto a la terminología de los subtipos del receptor alfa1-adrenérgico. Esto se debía al hecho de hablerse identificado subtipos diferentes utilizando técnicas farmacológicas y de biología molecular, si bien no estaba claro de qué manera estos subtipos diferentes se relacionaban entre sí. Con las técnicas farmacológicas se diferenciaban dos subtipos: alfa-1A y alfa-1B. Sin embargo, en los estudios de biología molecular fue posible clonar y expresar tres subtipos: alfa1b, alfa-1c y alfa-1d (denominados también alfa-1 a/d). Recientemente se ha aceptado que el receptor alfa1b-adrenérgico clonado codifica el receptor alfa 1B-adrenérgico farmacológico y que el receptor alfa1c-adrenérgico clonado alfa-1d también ha sido recientemente identificado mediante técnicas farmacológicas.
Varios estudios han demostrado que es el receptor alfa-1A-adrenérgico (designado previamente como el receptor alfa-1c-adrenérgico clonado) el que predomina tanto numéricamente (aproximadamente 70% de todos los receptores alfa-1-adrenérgicos) como funcionalmente en la próstata humana. Hasta ahora, la investigaciónha prestado poca atención a la cuestión de cuál de los receptores alfa-1-adrenérgicos es responsable de la contracción del músculo liso en los vasos sanguíneos y, por consiguiente de la regulación de la presión arterial en humanos. Un estudio reciente demuestra que en las grandes arterias humanas, como es la arteria ilíaca interna, el subtipo del receptor adrenérgico alfa-1B es probablemente el responsable de la vasoconstricción. Los autores concluyen que un antagonista del receptor adrenérgico alfa-1 con baja afinidad para el receptor adrenérgico alfa-1B puede lograr una respuesta terapéutica a la obstrucción de la salida vesical. Con menos efectos secundarios. Sin embargo este punto todavía tiene que ser confirmado con nuevas investigaciones. Se anticipa que los bloqueantes alfa-1 que antagonizan todos los subtipos del receptor alfa-1-adrenérgico reducen tanto la resistencia uretral como la presión arterial. Ninguno de los bloqueante alfa-1 actualmente comercializados (prazosina, alfuzosina, terazosina y doxazosina) se consideran específicos para cualquiera de los subtipos del receptor alfa-1.adrenérgico. Por el contrario, varios estudios han demostrado que la tamsulosina es específica para el receptor adrenérgico alfa-A. la tamsulosina es aproximadamente 12 veces más selectiva para los receptores alfa-1-adrenérgicos que están presentes en la próstata humana que los que se encuentran en la aorta humana. En un estudio reciente, la tamsulosina demostró una afinidad aproximadamente 20 veces más elevada hacia el subtipo alfa-1a clonado que hacia el subtipo clonado que hacia el subtipo de receptor alfa-1b-adrenérgico clonado. Ninguno de los alfa-bloquantes anteriores estudiados demostró una clara selectividad hacia el subtipo alfa-1a. *Clínica: En un amplio programa de ensayos clínicos, el primer antagonista específico de subtipo alfa-1A, tamsulosina, ha demostrado una eficacia comparable a la de los antiguos alfa-1-bloqueantes, demostrándose la esperada rápida mejoría de los síntomas de la HPB. Se observó que tamsulosina mejora significativamente los síntomas de HPB y scores de calidad de vida (QOL) después de 1 semana de tratamiento. Además, los efectos de tamsulosina sobre los Qmáx fueron vistos en el primer día de tratamiento con 0.4 mg de tamsulosina. El porcentaje de pacientes que alcanzó una mejoría clínicamente significativa de los síntomas fue aproximadamente el 70%, que mantuvo durante 12 meses de tratamiento. 0.4 mg de tamsulosina 1 vez al día carece de efecto clínicamente significativo sobre la presión arterial en posición supina o bipedestación y sobre la frecuencia de pulso en comparación en el efecto de placebo. Esto vale tanto para pacientes HPB hipertensos como normotensos. En una comparación directa, 2.5 mg de alfuzosina 3 veces al día produjeron significativamente mayores reducciones de la presión arterial que 0.4 mg de tamsulosina 1 vez al día. Los resultados de estos estudios indican que la tamsulosina puede administrarse concomitantemente, y sin necesidad de ajustes de la dosis, con varios fármacos que a menudo son administrados a los pacientes ancianos. El hecho de que la tamsulosina no interfiera con fármacos como atenolol, enalapril y nifedipina constituye una ventaja real en el manejo total del paciente anciando y ayuda a asegurar la calidad de vida en esta población de pacientes. La incidencia de eventos adversos en los pacientes tratados con 0.4 mg de tamsulosina 1 vez al día (sin titulación de dosis) no muestra diferencias estadísticamente significativas con las observadas en pacientes tratados con placebo (en los estudios controlados con placebo). Además, los eventos adversos comúnmente atribuidos a bloqueantes alfa-1 fueron observados con la misma frecuencia en los grupos placebo y tamsulosina 0.4 mg. En los estudios europeos y norteamericanos solo se observó mayor frecuencia en eyaculación retrógrada y rinitis en el grupo de tamsulosina 0.4 mg. Estos eventos adversos han sido comunicados con otros alfa-1-bloqueantes con una incidencia de 4-11% y están probablemente relacionados con su mecanismo de acción. Estos datos clínicos respaldan la hipóteiss de que con el bloqueo selectivo de los receptores alfa-1A-adrenérgicos puede alcanzarse un perfil de seguridad favorable. Destino en el organismo (farmacocinética): Absorción y biodisponibilidad: El clorhidrato de tamsulonsina se absorbe de forma gradual y casi totalmente desde el intestino después de una dosis oral de 0.4 mg. Su biodisponibilidad es casi del 100% (basado en el cociente del área bajo la curva [AUC] de una dosis única de 0.4 mg y una dosis intravenosa de 0.125 mg administrada por infusión a lo largo de 4 horas). Esto indica que el metabolismo de primer paso es despreciable. Las concentraciones plasmáticas en estado estable se alcanzan al quinto día después de una dosificación múltiple con 0.4 mg de tamsulosina 1 vez al día. La amplitud y tasa de la absorción queda reducida por una comida reciente (la AUC se reduce aproximadamente en un 30%; el tiempo hasta la concentración plasmática máxima [Tmax] se prolonga en aproximadamente 1 hora). El propio paciente puede mejorar la uniformidad de la absorción al tomar siempre la tamsulosina después del desayuno. En estas condiciones, el Tmax es de aproximadamente 6 horas. El Tmax es más prolongado para tamsulosina (formulación en liberación controlada) que para los demás alfa-1-bloqueantes. Esto, además de su selectividad alfa-1A, puede ser beneficioso para el perfil de seguridad de tamsulosina. En 13 pacientes ancianos (≥65 ańos) con HPB que recibieron 0.4 mg de tamsulosina 1 vez al día durante 8 días, el promedio de concentración plasmática máxima en estado estable (Cmax) en condiciones de ayuno fue de 17 ng/ml. Se produjo una variación intersujetos en los niveles plasmáticos, oscilando la Cmax entre 6 y 48 ng/ml. En 2 pacientes, sus elevados valores de Cmax (37 y 48 ng/ml) no se asociaron con ningún evento adverso. En estos pacientes ancianos HPB (edad media 73 ańos), la tasa de absorción estaba reducida (el Tmax medio fue de 11 horas). Distribución y ligadura a proteínas plasmáticas: La tamsulosina se liga altamente (aproximadamente en un 90%) a proteínas plasmáticas. Se fija principalmente a la glicoproteína ácida alfa-1, un componente de globulina del plasma, cuya concentración aumenta con la edad. El volumen de distribución de tamsulosina es reducido (aproximadamente 0.2 litro/kg). Metabolismo: La tamsulosina es maetabolizada lentamente por el hígado. Eliminación: La tamsulosina experimenta aclaramiento restrictivo. La mayoría de los metabolitos de tamsulosina tienen una alta afinidad por los receptores alfa-1-adrenérgicos, similar a la de la droga madre, y retiene su especificidad relativa para los receptores alfa-1A - adrenérgicos comparado con los alfa-1B. Los metabolitos de tamsulosina son rápidamente eliminados del plasma. La mayor parte de la tamsulosina está presente en el plasma en forma de fármaco inalterado. La tamsulosina y sus metabolitos se excretan principalmente en la orina (alrededor del 76%) encontrándose aproximadamente el 9% de la dosis en forma de fármaco inalterado. El aclaramiento sistémico medio de la tamsulosina es bajo: aproximadamente 48 ml/min. La vida media de eliminación (t medio) en pacientes ancianos con HPB en condiciones de estado estable es de aprox. 13 horas. La vida media terminal es de aproximadamente 22 horas. Esta prolongada vida media de tamsulosina en la formulación de liberación controlada permite utilizar un esquema podológico de una toma diaria. Efecto de la enfermedad sobre la farmacocinética: Insuficiencia renal: Se administró una dosis oral única de 0.4 mg de tamsulosina a 9 sujetos con grados variables de deterioro renal (aclaramiento de creatinina CLcr ≤70 ml/min) y a 5 sujetos con función renal normal (CLcr ≤90 ml/min). En 4 de los 9 sujetos, la función renal estaba severamente reducida (CLcr 10-30 ml/min) y en 5 estaba moderadamente reducida (CLcr 30-70 ml/min). Los grupos se compararon en relación a parámetros farmacocinéticos (por ej., Cmáx, Tmáx, tiempo medio, AUC, aclaramiento total aparente [CL/F], aclaramiento hepático intrínseco aparente [Clint/F] y fracción de fármaco no ligado en plasma). En pacientes con función renal moderadamente deteriorada, el tiempo medio aumentó de forma estadísticamente significativa en un 71% y el Clint/F descendió de forma estadísticamente significativa en un 50% en comparación con los sujetos con función renal normal. La reducción en el Clint/F fue influenciada por la edad; la edad media en pacientes con función renal moderadamente deteriorada era de 64.2 ańos, mientras que en los pacientes con insuficiencia renal severa y función renal normal era de 47.0 ańos y 51.5 ańos, respectivamente. Adicionalmente, la fracción de fármaco no ligado en el plasma fue un 18% menor en los pacientes con función renal moderadamente deteriorada que en el grupo con función renal normal. Esto estaba causado por una concentración más elevada de glicoproteína ácida alfa-1 en el grupo de sujetos con deterioro moderado, lo que se debía, probablemente, a la mayor edad de estos sujetos. Esta fracción más pequeńa de fármaco no ligado disminuirá el aclaramiento plasmático del fármaco y prolongará el tiempo medio. Dado que tamsulosina experimenta un aclaramiento restrictivo, la menor eliminación en el grupo con función renal moderadamente deteriorada se relaciona con la fracción de fármaco no ligado en el plasma y el aclaramiento hepático intrínseco aparente (ambos disminuyen con la edad) y no directamente con la función renal deteriorada. Por consiguiente, no es necesario realizar ajuste de dosis en los pacientes con insuficiencia renal. Insuficiencia hepática: Se administró una dosis oral única de 0.4 mg de tamsulosina a 8 sujetos con insuficiencia hepática de leve a moderada y 8 sujetos con función hepática normal. Los parámetros farmacinéticos evaluados fueron los mismos que en el estudio de insuficiencia renal. Como podía esperarse, había un descenso en la concentración de las 2 principales proteínas plasmáticas sintetizadas por el hígado (albúmina y glicoproteína ácida alfa-1), en los sujetos con insuficiencia hepática. El descenso en la glicoprotína ácida alfa-1 fue el principal responsable del aumento estadísticamente significativo del 150% en la fracción de tamsulosina no ligada en el plasma de los pacientes del grupo con insuficiencia hepática en comparación con los sujetos normales. Por consiguiente, aunque el aclaramiento hepático intrínseco aparente [Clint/F] evidentemente descendió en los sujetos con insuficiencia hepática, el aclaramiento total aparente [CL/F] aumentó ligeramente pero no de forma estadísticamente significativa. No obstante, dado que el volumen de distribución aparente también aumentó (aunque no de forma estadísticamente significativa), el cambio en la vida media de eliminación no fue estadísticamente significativo. En consecuencia, en pacientes con insuficiencia hepática leve o mederada, la tamsulosina puede administrarse a la dosis usual de 0.4 mg 1 vez al día. Por lo tanto, no es necesario realizar ajuste de dosis.
Posología: Una cápsula una vez al día a tomar después del desayuno. La cápsula deberá ser tragada entera con un poco de agua (alrededor de 15 ml) estando en posición de pie o sentado. Las cápsulas no deben aplastarse ni masticarse, ya que esto interferiría con la liberación controlada del principio activo.
Efectos Colaterales:Durante el uso de tamsulosina se han comunicado las siguientes reacciones adversas: mareos, eyaculación anormal y, con menos frecuencia (1-2%), cefaleas, astenia, hipotensión postural y palpitaciones.
Contraindicaciones: Hipersensibilidad a cualquiera de los componentes del producto. Hipotensión ortostática. Insuficiencia hepática severa. Durante la terapia con tamsulosina puede producirse un descenso en la presión arterial que, en muy raras ocasiones, puede provocar un síncope. A los primeros signos de hipotensión ortostática (mareo, debilidad); el paciente deberá sentarse o acostarse hasta que los síntomas hayan desaparecido. Antes del tratamiento, y a intervalos regulares a partir del inicio del mismo, deberá realizarse un tacto rectal y una determinación del antígeno prostático específico.
Precauciones: Deben excluirse otras afecciones de próstata que puedan causar síntomas similares a la hipertrofia benigna de próstata antes de iniciar el tratamiento con tamsulosina. Efectos sobre la capacidad para conducir y utilizar maquinaria: No existen datos disponibles sobre si tamsulosina afecta adversamente la capacidad para conducir o utilizar máquinas. Sin embargo, en este aspecto los pacientes deberán ser conscientes del hecho de que puede producir mareos.
Interacciones Medicamentosas: No se han observado interacciones al administrar tamsulosina conjuntamente con atenolol, enalapril o nefedipina. La administración concomitante de cimetididna produce una elevación de los niveles plasmáticos de tamsulosina, y furosemida una descenso, pero siempre que los niveles se mantengan dentro del rango posológico normal no es necesario realizar cambios. Ni diazepam ni propanolol, triclormetiazida, clormadinona, amitriptilina, diclofenac, glibenclamida, simvastaina y warfarina modifican la fracción libre de tamsulosina en el plasma humano. Tampoco tamsulosina cambia las fracciones libres de diazepam, propanolol y triclormetiazida. No se han observado interacciones al nivel del metabolismo hepático ligado al citocromo P450 que involucra a amitriptilina, salbutamol, glibenclamida y finasteride. El diclofenaco y la warfarina, sin embargo, pueden incrementar la tasa de eliminación de tamsulosina. La administración simultánea de otros antagonistas del receptor alfa-1 adrenérgico puede aumentar el riesgo de producir hipotensión.
Sobredosificación:Síntomas y tratamientos en dosis excesivas: No se han comunicado casos de sobredosis aguda. No obstante, después de una sobredosis, teóricamente, puede producirse hipotensión aguda, en cuyo caso deberá administrarse sostén cardiovascular. Deberá reestablecerse la presión arterial devolviendo la frecuencia cardíaca a la normalidad haciendo que el paciente se acueste. Si esto no sirve de ayuda, entonces deberán emplearse expandidores de volumen y, cuando sea necesario, vasopresores. Deberán monitorizarse la función renal y aplicarse medidas de apoyo general. Es improbable que la diálisis sirva de ayuda, ya que la tamsulosina se fija muy altamente a protínas plasmáticas. Pueden tomarse medidas, como por ejemplo, emesis para impedir la absorción. Cuando están involucradas grandes cantidades, puede aplicarse un lavado gástrico y puede administrarse carbón activado y un laxante osmótico, por ejemplo, sulfato sódico.
Conservación: En su envase original, a temperatura ambiente (15ş-30şC), al abrigo de la luz y la humedad excesiva.
 Laboratorio
Laboratorio