Composición: Cada comprimido contiene: 50 mg de Lamotrigina. Excipientes c.s.: Lactosa Monohidrato, Celulosa Microcristalina, Oxido de Hierro, Amarillo, Povidona, Almidón Glicolato de Sodio, Talco, Estearato de Magnesio.
Acción Terapéutica:Grupo farmacológico: Otros antiepilépticos. Código ATC: N03AX09.
Indicaciones:Daksol® está indicado en el tratamiento de: Epilepsia: Su uso combinado está indicado como terapia adjunta en crisis parciales en pacientes adultos y pediátricos de 2 ańos de edad o mayores. También está indicado como terapia adjunta en las crisis generalizadas, incluidas las convulsiones tónico-clónicas y las asociadas al síndrome de Lennox-Gastaut en pacientes adultos y pediátricos de 2 ańos de edad o mayores. En monoterapia está indicado para la conversión a monoterapia en pacientes adultos con crisis parciales o crisis generalizadas, incluidas las convulsiones tónico-clónicas y las asociadas al síndrome de Lennox-Gastaut y que están en tratamiento con un solo medicamento anticonvulsivante inductor enzimático. La seguridad y la eficacia de lamotrigina no han sido establecidas en los siguientes casos: 1. Como monoterapia inicial. 2. Para conversión a monoterapia a partir del tratamiento con un anticonvulsivante no inductor enzimático (ej: valproato). 3. Para conversión simultánea a monoterapia a partir de un tratamiento previo con 2 o más medicamentos anticonvulsivantes. 4. En pacientes menores de 16 ańos, a excepción de aquellos con crisis parciales y crisis generalizadas asociadas al síndrome de Lennox-Gastaut. En trastorno bipolar, adultos de 18 ańos y más de edad: está indicado para el tratamiento de mantención del desórden bipolar tipo I para disminuir la frecuencia de aparición de trastornos del ánimo (depresión, manía, hipomanía, episodios mixtos). Lamotrigina no está indicado para el tratamiento agudo de episodios maníacos o depresivos.
Propiedades:Propiedades farmacodinámicas: En pruebas diseńadas para evaluar los efectos de los fármacos sobre el sistema nervioso central, los resultados obtenidos empleando dosis de 240 mg de lamotrigina administrada a voluntarios sanos adultos, no se diferenciaron de los obtenidos empleando placebo, en tanto que, 1000 mg de fenitoína y 10 mg de diazepam alteraron notablemente, entre la coordinación fina visual motora y movimientos oculares, incrementaron el desequilibrio corporal y produjeron efectos sedativos subjetivos. En otro estudio, dosis única por vía oral de 600 mg de carbamazepina, alteraron significativamente la coordinación fina motora visual y movimientos oculares incrementando tanto el desequilibrio corporal como la frecuencia cardíaca, mientras que los resultados obtenidos con dosis de lamotrigina de 150 y 300 mg no se diferenciaron de los resultados obtenidos con placebo. Mecanismo de acción: los resultados de los estudios farmacológicos sugieren que Daksol® es un bloqueador de los canales de sodio voltaje dependientes. Produce un bloqueo, uso y voltaje dependiente, de neuronas cultivadas hiperexcitadas repetidamente e inhibe la liberación patológica del glutamato (aminoácido que juega un papel clave en la generación de crisis epilépticas), así como los potenciales de acción evocados tras la administración de glutamato. Propiedades farmacocinéticas: Absorción: Daksol® se absorbe completamente y rápidamente en el intestino con un insignificante metabolismo de primer paso. El pico de concentraciones plasmáticas se produce aproximadamente 1.5 horas tras la administración oral del fármaco. El tiempo hasta conseguir la concentración máxima se retrasa ligeramente después de ingerir alimento aunque la extensión de la absorción no se ve afectada. El perfil farmacocinético es lineal hasta 450 mg, la dosis más alta examinada. Existe una considerable variación interindividual en las concentraciones máximas en estado de equilibrio estacionario, pero dentro del mismo individuo las concentraciones varias muy poco. Distribución: el volumen de distribución es 0.92 a 1.22 l/kg. La unión a proteínas plasmáticas es de alrededor del 55%; es muy poco probable que el desplazamiento de las proteínas pueda dar lugar a toxicidad. La vida media de Daksol® se redujo aproximadamente a 14 horas cuando se administró con fármacos inductores enzimáticos, tales como carbamazepina y fenitoína, y se incrementa a una media de 70 horas aproximadamente cuando se coadministra únicamente con valproato sódico. Metabolismo: Daksol® induce su propio metabolismo en una modesta extensión dependiendo de la dosis. Sin embargo, no existe evidencia de que Daksol® afecte la farmacocinética de otros fármacos antiepilépticos y los datos sugieren que las interacciones entre Daksol® y fármacos metabolizados por enzimas del citocromo P-450 son poco probables. Eliminación: el clearance medio en el estado de equilibrio estacionario en adultos sanos es 39+14 ml/min. El clearance de Daksol® es primariamente metabólico con eliminación posterior del glucurónido conjugado en orina. Menos del 10% se excreta inalterado en orina. Solo el 2% del material relacionado con el fármaco se excreta en heces. El clearance y la vida media son independientes de la dosis. La vida media de eliminación en adultos sanos es 24 a 35 horas. Las UDP-glucoroniltransferasas han sido identificadas como las enzimas responsables del metabolismo de Daksol®. En un estudio de sujetos con el síndrome de Gilbert, el clearance medio aparente se redujo en un 32% comparado con los controles normales aunque los valores estaban dentro del rango para la población general. El clearance ajustado al peso corporal es mayor en nińos que en adultos, con los valores más altos en nińos de edad inferior a 5 ańos. La vida media de Daksol® es generalmente más corta en los nińos que en los adultos con un valor medio de aproximadamente 7 horas cuando se administra con fármacos inductores enzimáticos, tales como carbamazepina y fenitoína y se incrementa a valores medios de 45 a 50 horas cuando se coadministra únicamente con valproato sódico. Hasta la fecha no se han realizado estudios específicos de la farmacocinética de Daksol® en pacientes ancianos con epilepsia. Sin embargo, un estudio con dosis en 12 voluntarios sanos de edades comprendidas entre 65 y 76 ańos y un análisis de una población de 144 pacientes incluyendo 25 pacientes de más de 65 ańos de edad, indicó que no es necesario realizar ajustes de dosis en el tratamiento de los ancianos. No existe experiencia en el tratamiento con Daksol® de pacientes con falla renal. Los estudios farmacocinéticos que utilizan dosis únicas en sujetos con falla renal indican que la farmacocinética de Daksol® se ve poco afectada, aunque las concentraciones plasmáticas del principal metabolito glucurónido se incrementan casi 8 veces debido a la reducción del clearance renal.
Posología:Epilepsia: Adultos (mayores de 12 ańos): Monoterapia: la dosis inicial de Daksol® es de 25 mg/día (1 vez al día), durante 2 semanas, seguida de 50 mg/día (1 vez al día), durante 2 semanas. Mantención: para conseguir la respuesta óptima es de 100-200 mg/día administrada 1 vez al día o en 2 dosis divididas. Algunos pacientes han precisado 500 mg/día de Daksol® para conseguir la respuesta deseada. No debe sobrepasarse la dosis inicial recomendada. Terapia ańadida: Adultos y nińos mayores de 12 ańos: la dosis incial de Daksol® en aquellos pacientes que no estén en tratamiento con valproato sódico es de 50 mg 1 vez al día, durante las 2 primeras semanas, seguida de 100 mg/día administrados en 2 tomas durante las 2 semanas siguientes. La dosis normal de mantenimiento es de 200-400 mg/día repartida en 2 tomas. Para los pacientes que estén tomando valproato sódico, la dosis inicial de Daksol® es de 25 mg en días alternos durante las 2 primeras semanas. Durante las 2 semanas siguientes, la dosis será de 25 mg al día. La dosis normal de mantenimiento es de 100 a 200 mg 1 vez al día o repartida en 2 tomas. En cualquier caso no se deberá exceder la dosis inicial recomendada. Nińos (2-12 ańos): con el fin de asegurar que se mantiene la dosis terapéutica, debe monitorizarse el peso del nińo. Si se producen cambios en el peso, la dosis debe ser revisada. Si las dosis calculadas para los nińos, de acuerdo al peso corporal no equivalen a comprimidos enteros, la dosis administrada debe ser igual al número más bajo de comprimidos.
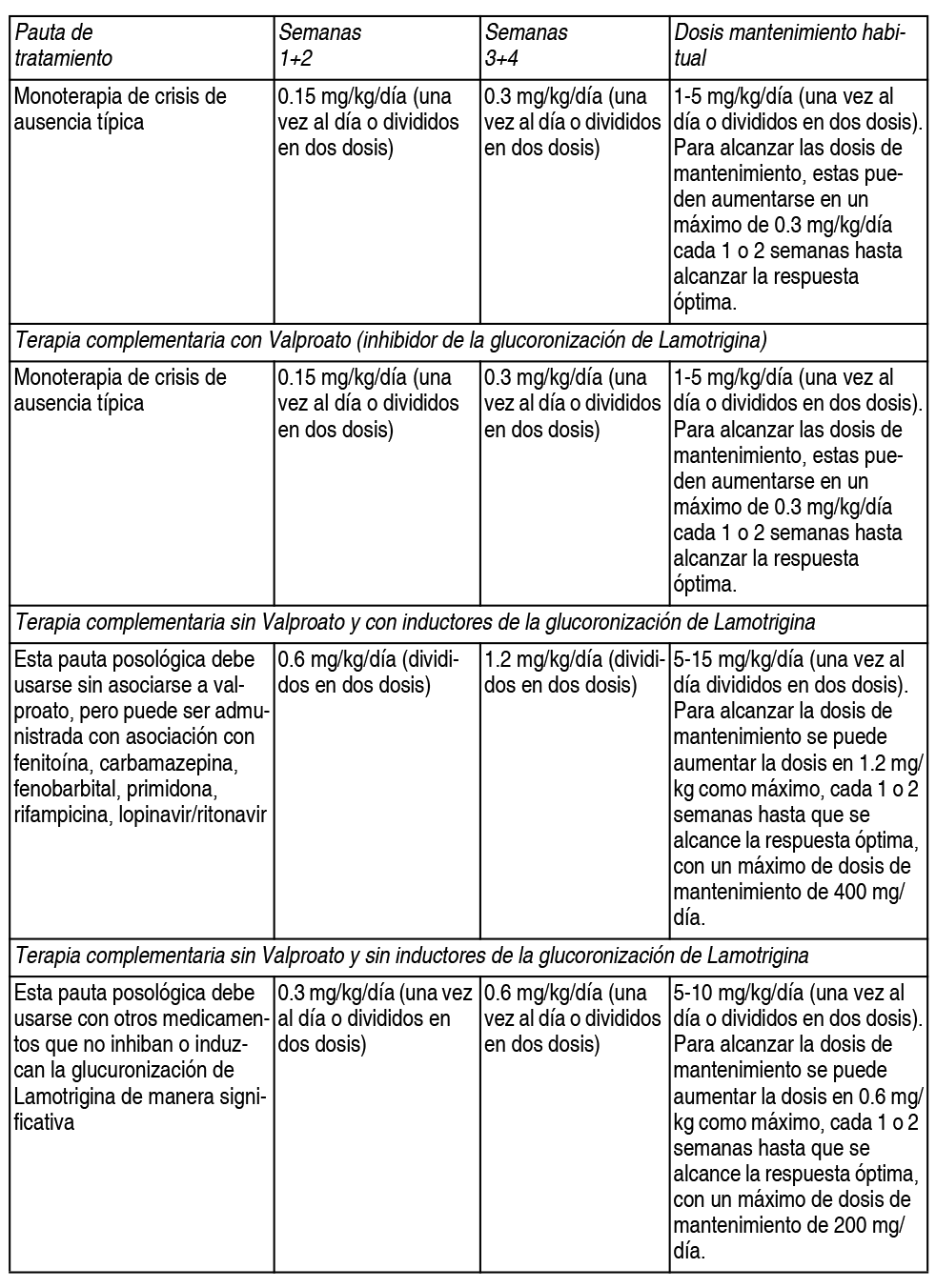
En pacientes que toman fármacos antiepilépticos de los que se desconoce la interacción farmacocinética con lamotrigina, se deben utilizar las dosis escalantes recomendadas cuando se administra Daksol® conjuntamente con valproato. Se debe controlar el peso del nińo para asegurar que se mantiene la dosis terapéutica y en caso de que se produzcan cambios en el peso del paciente, la dosis debe ajustarse. Si la dosis diaria calculada es de 2.5 - 5 mg, se puede tomar Daksol® 5 mg en días alternos durante las 2 primeras semanas. Si la dosis diaria calculada es menor de 2.5 mg no se debe administrar Daksol®. Debido al riesgo de erupción cutánea, no se debe exceder la dosis inicial ni la dosis escalante posterior. Es probable que los pacientes de edad comprendida entre 2-6 ańos necesiten una dosis de mantenimiento que sea la más alta del rango recomendado. Una vez se haya alcanzado el control epiléptico con el tratamiento complementario, los fármacos antiepilépticos administrados de forma concomitante pueden ser retirados y los pacientes pueden continuar en monoterapia con lamotrigina. Nińos menores de 2 ańos: no existe información suficiente sobre el uso de Daksol® en nińos menores de 2 ańos. Por lo tanto, no se recomienda el uso de lamotrigina en nińos menores de 2 ańos. Uso en ancianos: No se requiere modificar las dosis con respecto a la pauta recomendada. La farmacocinética de lamotrigina en este grupo de edad no varía significativamente con relación a la población adulta no anciana. Trastorno bipolar: Adultos (18 ańos de edad y mayores): debido al riesgo de exantema no deben excederse la dosis inicial ni el escalamiento subsecuente de la dosis (ver advertencias y precauciones especiales de uso). Lamotrigina se recomienda para uso en los pacientes con trastorno bipolar en riesgo de un futuro episodio depresivo. Para prevenir la recurrencia de los episodios depresivos se debe observar el siguiente régimen de transición: el régimen de transición involucra el escalamiento de la dosis de lamotrigina a una dosis de mantenimiento de la estabilización durante 6 semanas, después de lo cual, si está clínicamente indicado, otros fármacos psicotrópicos y/o antiepilépticos pueden ser discontinuados. Se debe tomar en cuenta la terapia adjunta para la prevención de episodios maníacos, ya que no ha establecido concluyentemente la eficacia de lamotrigina en la manía. a) terapia adjunta con inhibidores de la glucoronidación por ej. valproato. En pacientes que toman fármacos inhibidores de la glucoronidación concomitantes como valproato, la dosis inicial de lamotrigina es 25 mg cada día alterno por 2 semanas, seguidos de 25 mg 1 vez por día por 2 semanas. La dosis se debe aumentar hasta 50 mg 1 vez al día (o en 2 dosis divididas) en la semana 5. La dosis usual deseada para lograr la respuesta óptima es 100 mg/día administrados 1 vez al día o en 2 dosis divididas. Sin embargo, se puede aumentar la dosis hasta una dosis diaria máxima de 200 mg, dependiendo de la respuesta clínica. b) Terapia adjunta con inductores de la glucuronidación de lamotrigina en pacientes que no toman inhibidores como el valproato. Este régimen posológico se debe usar con fenitoína, carbamazepina, fenobarbitona, primidona y otros fármacos reconocidos por inducir la glucuronidación de lamotrigina. En los pacientes que actualmente toman fármacos que inducen la glucuronidación de lamotrigina y no toman valproato, la dosis inicial de lamotrigina es 50 mg 1 vez al día por 2 semanas. La dosis se debe aumentar hasta 200 mg/día administrados como 2 dosis divididas en la semana 5. En la semana 6, se puede aumentar la dosis a 300 mg/día sin embargo, la dosis usual deseada para lograr la respuesta óptima es 400 mg/día administrados en 2 dosis divididas que se pueden administrar a partir de la semana 7. c) Monoterapia con lamotrigina o terapia adjunta en pacientes que toman otros medicamentos que no inhiben ni inducen significativamente la glucoronidación de lamotrigina. La dosis inicial de lamotrigina, es de 25 mg 1 vez al día durante 2 semanas, seguida por 50 mg 1 vez (o dividido en 2 tomas diarias) durante 2 semanas. La dosis se debe aumentar hasta 100 mg/día en la semana 5. La dosis usual deseada para lograr la respuesta óptima es 200 mg/día administrados 1 vez al día o como 2 dosis divididas. Sin embargo, en las pruebas clínicas se usó un rango de 100 mg a 400 mg. Nińos y adolescentes (menores de 18 ańos de edad): el uso de lamotrigina no se indica en el trastorno bipolar en nińos y adolescentes menores de 18 ańos de edad. No se han evaluado la seguridad y la eficacia de lamotrigina para el trastorno bipolar en este grupo de edad. Por lo tanto, no se pueden hacer recomendaciones posológicas. Recomendaciones posológicas generales para lamotrigina en población de pacientes especiales: mujeres que toman anticonceptivos hormonales a) Inicio de lamotrigina en pacientes que ya toman anticonceptivos hormonales: aunque se ha demostrado que el anticonceptivo oral aumenta la eliminación de lamotrigina, no serán necesarios ajustes a las guías de escalamiento de dosis recomendadas para lamotrigina solamente con base en el uso de anticonceptivos hormonales. El escalamiento de la dosis deberá seguir las guías recomendadas con base en si se agrega lamotrigina a valproato (un inhibidor de la glucuronidación de lamotrigina) o a un inductor de la glucuronidación de lamotrigina o si se agrega lamotrigina en ausencia de valproato, o de algún inductor de la glucuronidación de la lamotrigina. b) Inicio de anticonceptivos hormonales en pacientes que ya toman la dosis de mantenimiento de lamotrigina y no toman inductores de la glucuronidación de lamotrigina. En la mayoría de los casis será necesario aumentar la dosis de mantenimiento de lamotrigina hasta el doble de acuerdo con la respuesta clínica individual. c) Interrumpiendo el uso de anticonceptivos hormonales en pacientes que ya estén tomando dosis de mantenimiento de lamotrigina y que no estén tomando sustancias inductoras de la glucuronidación de la lamotrigina: puede ser necesario disminuir la dosis de mantenimiento de la lamotrigina hasta un 50% de acuerdo con la respuesta individual.
Efectos Colaterales: Se han informado reacciones adversas cutáneas, que han aparecido generalmente en las primeras 8 semanas después de iniciado el tratamiento con Daksol®. La mayoría de las erupciones cutáneas son leves y autolimitantes, sin embargo, se han informado reacciones cutáneas graves, de riesgo potencial para la vida, que incluyen síndrome de Stevens-Johnson (SJS) y necrólisis tóxica epidérmica (síndrome de Lyell). En nińos, la aparición inicial de erupción cutánea se puede confundir con la infección, los médicos deben considerar la posibilidad de una reacción al fármaco en los nińos que desarrollen síntomas de erupción cutánea y fiebre durante las 8 primeras semanas del tratamiento. Adicionalmente, el riesgo global de aparición de la erupción está altamente asociado con: Dosis iniciales elevadas de Daksol® que exceden la dosis escalante recomendada en el tratamiento con lamotrigina. Uso de valproato conjunta, ente, el cual incrementa la vida media de Daksol® casi a valores duplicados. Todos los pacientes (adultos y nińos) que desarrollen erupción deben ser evaluados rápidamente. Se debe retirar el tratamiento con Daksol® inmediatamente a menos que claramente, la erupción no esté relacionada con el fármaco. La erupción se ha informado como parte del síndrome de hipersensibilidad asociado con un modelo variable de síntomas sistémicos que incluyen fiebre, linfadenopatía, edema facial, anomalías en la sangre e hígado. El síndrome muestra un amplio espectro de gravedad clínica y puede, raramente conducir a coagulación intravascular diseminada (CID) y fallo multiorgánico. Es importante remarcar que incluso cuando la erupción no es evidente, pueden aparecer manifestaciones tempranas de hipersensibilidad (ej. Fiebre, linfadenopatía). Si aparecen tales signos y síntomas, el paciente tiene que ser evaluado inmediatamente y suspender Daksol® si no se puede establecer una etiología alternativa. Como en el caso de otros fármacos antiepilépticos, la retirada brusca de Daksol® puede provocar efecto rebote. La dosis de Daksol® debe reducirse de forma progresiva durante un período de 2 semanas, a menos que existan aspectos de seguridad (ej. Erupción) que requieran una retirada brusca del producto. Cuando se retiran fármacos antiepilépticos concomitantes para alcanzar la monoterapia con Daksol® o se ańaden otros fármacos antiepilépticos a la monoterapia con Daksol® debe tenerse en cuenta el efecto que puede tener sobre la farmacocinética de Daksol®. Daksol® es un débil inhibidor de la dihidrofolato reductasa y, por lo tanto, existe una posibilidad de interferencia con el metabolismo del folato a lo largo de un tratamiento de larga duración. De todas formas en el tratamiento prolongado en humanos de hasta 1 ańo de duración, Daksol® no indujo cambios significativos en la concentración de hemoglobina, volumen corpuscular medio y concentraciones de folato en suero o en glóbulos rojos. La principal vía de eliminación del Daksol® es el metabolismo hepático, seguido de excreción renal. Hasta que se hayan realizado estudios específicos, no se puede recomendar actualmente el empleo de Daksol® en pacientes con una alteración significativa de las funciones renal o hepática. Cuando otros fármacos antiepilépticos se ańaden a la monoterapia con Daksol®, debe tenerse en consideración el efecto que ello pueda tener sobre la farmacocinética de Daksol®. Existen informes en la literatura referentes a que crisis convulsivas graves incluyendo status epilépticus pueden conducir a rabdomiólisis, disfunción multiorgánica y coagulación intravascular diseminada, con resultado fatal en algunas ocasiones. Se han producido casos similares en asociación con el uso de Daksol®. Empeoramiento clínico y riesgo de suicidio: es posible que se presenten síntomas de depresión y/o trastorno bipolar en pacientes con epilepsia, además existen indicios de que los pacientes con epilepsia y trastorno bipolar tienen un riesgo mayor de suicidalidad. Por lo tanto, se debe aconsejar a los pacientes (y a sus cuidadores) que busquen orientación médica si surgen signos de ideación o comportamiento suicidas. En pacientes con trastorno bipolar puede producirse un empeoramiento de los síntomas depresivos y/o tendencias suicidas emergentes, con independencia de que estén tomando medicación para el trastorno bipolar, incluyendo tratamiento con lamotrigina. Por lo tanto, se debe monitorizar estrechamente a aquellos pacientes que estén en tratamiento con lamotrigina para el trastorno bipolar y que presenten un empeoramiento clínico (incluyendo el desarrollo de nuevos síntomas) y tendencias suicidas. Esta monitorización es especialmente importante al inicio del tratamiento y cuando se realicen cambios/ajustes en la dosis. Algunos pacientes, incluyendo los que presentan un historial de comportamiento o pensamientos suicidas, adultos jóvenes y aquellos pacientes que presentan un grado significativo de ideación suicida anterior al inicio del tratamiento, pueden presentar un mayor riesgo de intentos y pensamientos suicidas. Por este motivo deben monitorizarse cuidadosamente durante el tratamiento. Se recomienda el cambio de régimen de tratamiento, incluyendo la posible discontinuación de la medicación, en pacientes que experimenten un empeoramiento clínico (incluyendo el desarrollo de nuevos síntomas) y/o ideación/comportamiento suicida emergente. Esto vale especialmente si estos síntomas son severos, de brusca aparición o sino formaban parte de los síntomas presentes en el paciente. Los acontecimientos adversos informados durante los ensayos con Daksol® en monoterapia incluyen dolor de cabeza, cansancio, erupción cutánea, náuseas, vértigo, somnolencia e insomnio. El riesgo global de aparición de la erupción está altamente asociado con: dosis iniciales elevadas de lamotrigina que exceden la dosis escalante recomendada en el tratamiento con lamotrigina. Uso de valproato concomitante, el cual incrementa la semivida de lamotrigina casi al doble. La erupción ha sido informada como parte del síndrome de hipersensibilidad asociado con un modelo variable de síndromes sistémicos que incluyen fiebre, linfadenopatía, edema facial y anomalías en la sangre e hígado. El síndrome muestra un amplio espectro de gravedad clínica y puede, raramente, conducir a coagulación intravascular diseminada (CID) y fallo multiorgánico. Es importante resaltar que incluso cuando la erupción no es evidente, pueden aparecer manifestaciones tempranas de hipersensibilidad (ej. Fiebre, linfadenopatía). Si tales signos y síntomas aparecen, el paciente tiene que ser evaluado inmediatamente y suspender Daksol® si no se puede establecer una etiología alternativa. Otros efectos adversos comunicados con Daksol® como terapia ańadida a otros regímenes estándar con fármacos antiepilépticos, incluyen diplopía, visión borrosa, conjuntivitis, mareo, aturdimiento, dolor de cabeza, inestabilidad, cansancio, alteración gastrointestinal (incluyendo vómitos), irritabilidad/agresividad, temor, agitación, anomalías hematológicas (incluyendo leucopenia y trombocitopenia) y confusión.
Contraindicaciones: Daksol® está contraindicado en individuos con conocida hipersensibilidad a lamotrigina. No se recomienda en insuficiencia renal o hepática. Se recomienda el monitoreo intensivo (incluyendo parámetros de coagulación hepáticos y renales) en pacientes que desarrollen en forma aguda cualquier combinación de rash inexplicado, fiebre, síntomas tipo gripales, o empeoramiento del control de las convulsiones. Evitar consumo de alcohol.
Precauciones:Efectos sobre la capacidad para conducir y utilizar máquinas: Tenga precaución antes de manejar un auto o alguna maquinaria hasta que este seguro de si lamotrigina afecta o no su habilidad para realizar estas tareas. Fertilidad, embarazo y lactancia: Embarazo: Se dispone de datos insuficientes relativos al empleo de Daksol® en el embarazo, para evaluar su seguridad. No se recomienda la administración de Daksol® durante el embarazo, a menos que en opinión del médico, el potencial beneficio del tratamiento para la madre compense cualquier riesgo para el feto en desarrollo. Lamotrigina es un débil inhibidor de la dihidrofolato reductasa. Existe un riesgo teórico de malformaciones fetales cuando la madre se trata con un inhibidor del folato durante el embarazo. El riesgo de malformaciones congénitas se ve incrementado de 2 a 3 veces en los recién nacidos de madres tratadas con fármacos antiepilépticos, en comparación con la incidencia aproximada del 3% esperada en la población general. Los defectos notificados en forma más frecuente son labio leporino, malformaciones cardiovasculares y defectos del tubo neural. La terapia con varios fármacos antiepilépticos se ha asociado a un mayor riesgo de malformaciones congénitas que el uso en monoterapia. Sin embargo los estudios de toxicología reproductiva realizados con lamotrigina en animales a dosis superiores a la dosis terapéutica en humanos, no mostraros efectos teratogénico. Lactancia: Se ha notificado que la lamotrigina pasa a la leche materna en concentraciones altamente variables, dando como resultado concentraciones totales de lamotrigina en lactantes de hasta aproximadamente 50% de las observadas en las madres. Por lo tanto, en algunos lactantes amamantados, las concentraciones séricas de lamotrigina podrían alcanzar niveles a los que se produzcan efectos farmacológicos. No se dispone de información sobre las concentraciones de lamotrigina o de sus metabolitos que pueden aparecer en la leche materna tras la administración de lamotrigina, por lo tanto no se recomienda la administración de lamotrigina a madres en período de lactancia.
Interacciones Medicamentosas:Interacción con otros medicamentos y otras formas de interacción: No se tiene evidencia de que Daksol® origine inducción o inhibición clínicamente significativa, de las enzimas responsables del metabolismo hepático oxidativo de fármacos. Daksol® puede inducir su propio metabolismo, pero el efecto es escaso con pocas probabilidades de presentar consecuencias clínicas significativas. Se han observado en algunos pacientes, incrementos en las concentraciones plasmáticas de otros antiepilépticos, aunque los estudios controlados no han presentado evidencia de que Daksol® afecte las concentraciones plasmáticas de fármacos antiepilépticos concomitantes. Las observaciones procedentes de estudios in vitro indican que Daksol® no desplaza a otros fármacos antiepilépticos concomitantes. Anticonceptivos orales: en un estudio realizado con 12 voluntarios, Daksol® no afectó las concentraciones plasmáticas de etinilestradiol y levonorgestrel tras la administración del anticonceptivo oral. De todas formas, como es el caso de la inducción de otro tratamiento crónico en pacientes tomando anticonceptivos orales, cualquier alteración en el perfil de hemorragia menstrual debería ser comunicada al médico de la paciente. Los agentes antiepilépticos como fenitoína, carbamazepina, fenobarbital y primidona que inducen las enzimas responsables del metabolismo hepático, aumentan el metabolismo de lamotrigina. Valproato sódico, inhibidor de la enzima responsable del metabolismo hepático, reduce el metabolismo de Daksol®. Carbamazepina: existen informes sobre el sistema nervioso central que incluyen vértigo, ataxia, diplopía, visión borrosa y náuseas en pacientes que toman carbamazepina, después de la introducción de Daksol®. Estos efectos se resuelven normalmente cuando se reduce la dosis de carbamazepina.
Sobredosificación:Signos y síntomas: se ha informado en un pequeńo número de pacientes la ingestión de entre 1.35 g y 4 g de lamotrigina. Las consecuencias clínicas incluyeron los signos y síntomas: nistagmo, ataxia, mareo y coma. Tratamiento: en caso de sobredosis, el paciente debe ser ingresado en un hospital y se le debe aplicar el tratamiento adecuado. Si estuviera indicado, debería realizarse un lavado gástrico.
Conservación: Mantener lejos del alcance de los nińos, mantener en su envase original, protegido del calor, la luz y la humedad a temperaturas inferiores a los 25° C. No usar este producto después de la fecha de vencimiento indicada en el envase. No repita el tratamiento sin consultar antes con el médico. No recomiende este medicamento a otra persona.
 Laboratorio
Laboratorio