Composición: Cada comprimido recubierto contiene: 120 mg de Etoricoxib. Comprimidos recubiertos oblongos, de color verde. Lista de excipientes c.s.: Celulosa Microcristalina, Fosfato Hidrógeno de Calcio Anhidro, Croscarmelosa Sódica, Estearato de Magnesio, Lactosa Monohidrato, Hipromelosa 2910/15 cP, Dióxido de Titanio.
Acción Terapéutica:Grupo farmacoterapéutico: Productos antiinflamatorios y antirreumáticos, no esteroideos, coxibs. Código ATC: M01AH05.
Indicaciones:Está indicado para: Tratamiento de la dismenorrea primaria. Alivio del dolor agudo. Dolor asociado a la artritis gotosa aguda en un período máximo de 8 días. La decisión de prescribir un inhibidor selectivo de la COX-2 debe estar basada en una evaluación de cada uno de los riesgos individuales del paciente.
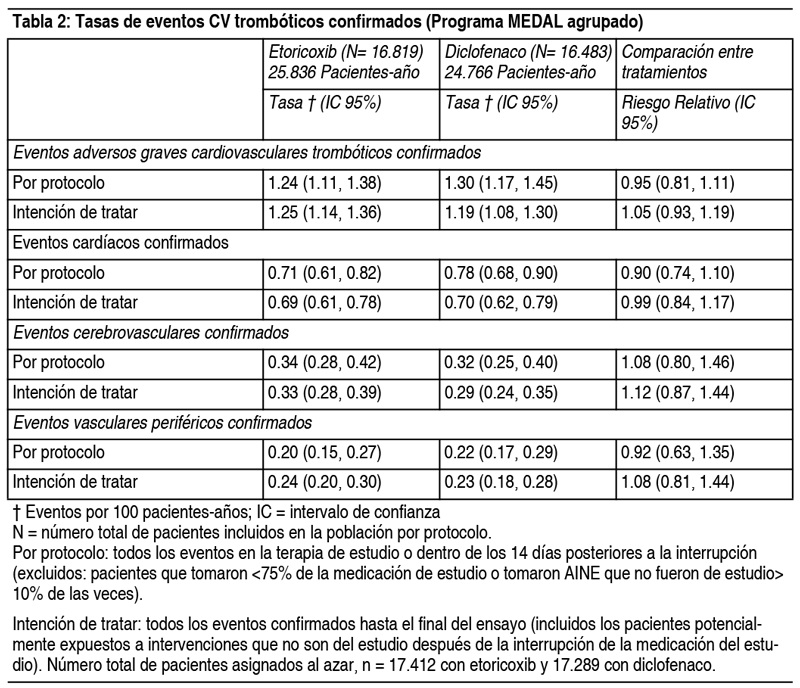
Propiedades:Propiedades farmacodinámicas: Mecanismo de acción: Etoricoxib es un inhibidor oral selectivo de la ciclooxigenasa 2 (COX-2) dentro del rango de dosis clínica. En todos los estudios de farmacología clínica, etoricoxib produjo una inhibición dependiente de la dosis de COX-2 sin inhibición de la COX-1 en dosis de hasta 150 mg al día. Etoricoxib no inhibió la síntesis de prostaglandinas gástricas y no tuvo efecto sobre la función plaquetaria. La ciclooxigenasa es responsable de la generación de prostaglandinas. Se han identificado 2 isoformas, COX-1 y COX-2. La COX-2 es la isoforma de la enzima que se ha demostrado que es inducida por estímulos proinflamatorios y se ha postulado que es la principal responsable de la síntesis de los mediadores prostanoides del dolor, la inflamación y la fiebre. La COX-2 también está involucrada en la ovulación, la implantación y el cierre del ductus arterioso, la regulación de la función renal y las funciones del sistema nervioso central (inducción de la fiebre, percepción del dolor y función cognitiva). También puede desempeńar un papel en la curación de la úlcera. La COX-2 ha sido identificada en el tejido alrededor de las úlceras gástricas en el hombre, pero su relevancia para la curación de la úlcera no ha sido establecida. Eficacia clínica y seguridad: Eficacia: En pacientes con osteoartritis (OA), etoricoxib 60 mg 1 vez al día proporcionó mejoras significativas en el dolor y las evaluaciones de los pacientes sobre el estado de la enfermedad. Estos efectos beneficiosos se observaron tan pronto como el segundo día de terapia y se mantuvieron durante hasta 52 semanas. Los estudios con etoricoxib 30 mg 1 vez al día demostraron una eficacia superior al placebo durante un período de tratamiento de 12 semanas (utilizando evaluaciones similares a las de los estudios anteriores). En un estudio de rango de dosis, etoricoxib 60 mg demostró una mejoría significativamente mayor que 30 mg para los 3 puntos finales primarios durante las 6 semanas de tratamiento. La dosis de 30 mg no se ha estudiado en la osteoartritis de manos. En pacientes con artritis reumatoide (AR), etoricoxib 90 mg 1 vez al día proporcionó mejoras significativas en el dolor, la inflamación y la movilidad. Estos efectos beneficiosos se mantuvieron durante los períodos de tratamiento de 12 semanas. En pacientes que sufren ataques de artritis gotosa aguda, etoricoxib 120 mg 1 vez al día durante un período de tratamiento de 8 días, alivió el dolor y la inflamación moderados a extremos de las articulaciones, comparables a la indometacina 50 mg 3 veces al día. Se observó alivio del dolor tan pronto como 4 horas después del inicio del tratamiento. En pacientes con espondilitis anquilosante, etoricoxib 90 mg 1 vez al día proporcionó mejoras significativas en el dolor, la inflamación, la rigidez y la función de la columna vertebral. El beneficio clínico de etoricoxib se observó tan pronto como el segundo día de tratamiento después del inicio del tratamiento y se mantuvo durante todo el período de tratamiento de 52 semanas. Seguridad: Programa multinacional Etoricoxib and Diclofenac Arthritis Long-term (MEDAL): El Programa MEDAL fue un Programa de Resultados de Seguridad Cardiovascular (CV) diseńado prospectivamente, de datos agrupados de 3 ensayos aleatorizados, comparativos, doble ciego y controlados, el estudio MEDAL, EDGE II y EDGE. Seguridad general: No hubo diferencias significativas entre etoricoxib y diclofenaco en la tasa de eventos trombóticos cardiovasculares. Los eventos adversos cardio-renales se observaron con mayor frecuencia con etoricoxib que con diclofenaco, y este efecto fue dosis-dependiente (ver resultados específicos a continuación). Los eventos adversos gastrointestinales y hepáticos se observaron significativamente más frecuentemente con diclofenaco que con etoricoxib. La incidencia de experiencias adversas en EDGE y EDGE II y de experiencias adversas consideradas serias o que dieron lugar a la interrupción en el estudio MEDAL fue mayor con etoricoxib que con diclofenaco. Resultados de seguridad cardiovascular: La tasa de eventos adversos graves cardiovasculares trombóticos confirmados (consistentes en eventos vasculares cardíacos, cerebrovasculares y periféricos) fue comparable entre etoricoxib y diclofenaco, y los datos se resumen en la tabla a continuación. No hubo diferencias estadísticamente significativas en las tasas de eventos trombóticos entre el etoricoxib y el diclofenaco en todos los subgrupos analizados, incluidas las categorías de pacientes en un rango de riesgo cardiovascular inicial. Cuando se consideran por separado, los riesgos relativos para los eventos cardiovasculares graves trombóticos confirmados con etoricoxib 60 mg o 90 mg en comparación con diclofenaco 150 mg fueron similares.
La mortalidad CV, así como la mortalidad global, fue similar entre los grupos de tratamiento con etoricoxib y diclofenaco. Resultados de la tolerabilidad gastrointestinal del programa MEDAL: Se observó una tasa significativamente menor de interrupciones del tratamiento para cualquier evento adverso gastrointestinal clínico (p. Ej., Dispepsia, dolor abdominal, úlcera) con etoricoxib en comparación con diclofenaco en cada uno de los 3 estudios de componentes del Programa MEDAL. Las tasas de interrupciones debidas a eventos adversos de GI clínico por 100 pacientes- ańo durante todo el período de estudio fueron las siguientes: 3.23 para etoricoxib y 4.96 para diclofenaco en el estudio MEDAL: 9.12 con etoricoxib y 12.28 con diclofenaco en el estudio EDGE; y 3,71 con etoricoxib y 4,81 con diclofenaco en el estudio EDGE II. Resultados de seguridad gastrointestinal del programa MEDAL: En general, los eventos GI superiores se definieron como perforaciones, úlceras y hemorragias. El subconjunto de eventos GI superiores en general considerados complicados incluyó perforaciones, obstrucciones y sangrado complicado; el subconjunto de eventos GI superiores considerados no complicados incluyó hemorragias no complicadas y úlceras no complicadas. Se observó una tasa significativamente menor de eventos GI superiores en general con etoricoxib en comparación con diclofenaco. No hubo diferencias significativas entre etoricoxib y diclofenaco en la tasa de eventos complicados. Para el subconjunto de eventos de hemorragia GI superior (combinado complicado y sin complicaciones), no hubo diferencias significativas entre etoricoxib y diclofenaco. El beneficio GI superior para etoricoxib comparado con diclofenaco no fue estadísticamente significativo en pacientes que tomaban aspirina en dosis baja concomitante (aproximadamente 33% de los pacientes). Las tasas por 100 pacientes-ańo de eventos clínicos GI superiores complicados y no complicados confirmados (perforaciones, úlceras y hemorragias [PUB]) fueron 0.67 (IC 95% 0.57, 0.77) con etoricoxib y 0.97 (IC 95% 0.85, 1.10) con diclofenaco, lo que arroja un riesgo relativo de 0,69 (IC del 95%: 0.57; 0.83). Se evaluó la tasa de eventos gastrointestinales superiores confirmados en pacientes de edad avanzada y se observó la mayor reducción en pacientes ≥75 ańos de edad (1.35 [IC 95% 0.94; 1.87] frente a 2.78 [IC 95% 2.14, 3.56] eventos por 100 pacientes -ańos para etoricoxib y diclofenac, respectivamente. Las tasas de eventos clínicos GI confirmados inferiores (perforación del intestino delgado o grande, obstrucción o hemorragia [POB]) no fueron significativamente diferentes entre el etoricoxib y el diclofenaco. Resultados de seguridad hepática del programa MEDAL: Etoricoxib se asoció con una tasa de discontinuidades estadísticamente significativamente más baja debido a las experiencias adversas relacionadas con el hígado que el diclofenaco. En el programa MEDAL combinado, el 0.3% de los pacientes con etoricoxib y el 2.7% de los pacientes con diclofenaco suspendieron debido a experiencias adversas relacionadas con el hígado. La tasa por 100 pacientes-ańos fue 0.22 con etoricoxib y 1.84 con diclofenaco (el valor de p fue <0.001 para etoricoxib vs. diclofenaco). Sin embargo, la mayoría de las experiencias adversas hepáticas en el Programa MEDAL no fueron graves. Propiedades farmacocinéticas: Absorción: Etoricoxib administrado por vía oral se absorbe bien. La biodisponibilidad absoluta es aproximadamente del 100%. Después de una dosis de 120 mg 1 vez al día hasta el estado estacionario, la concentración plasmática máxima (media geométrica Cmax = 3.6 µg/ml) se observó aproximadamente a la 1 hora (Tmax) después de la administración a adultos, en ayunas. El área media geométrica bajo la curva (AUC0-24hr) fue 37.8 µg•hr/ml. La farmacocinética de etoricoxib es lineal en todo el rango de dosis clínicas. La dosificación con alimentos (una comida rica en grasas) no tuvo ningún efecto sobre el grado de absorción de etoricoxib después de la administración de una dosis de 120 mg. La tasa de absorción se vio afectada, dando como resultado una disminución del 36% en Cmax y un aumento en Tmax de 2 horas. Estos datos no se consideran clínicamente significativos. En ensayos clínicos, etoricoxib se administró sin importar la ingesta de alimentos. Distribución: Etoricoxib se une aproximadamente en un 92% a la proteína plasmática humana en el rango de concentraciones de 0.05 a 5 µg/ml. El volumen de distribución en estado estacionario (Vdss) fue de aproximadamente 120 l en humanos. Etoricoxib atraviesa la placenta en ratas y conejos, y la barrera hematoencefálica en ratas. Biotransformación: Etoricoxib se metaboliza ampliamente con <1% de una dosis recuperada en la orina como fármaco original. La principal vía del metabolismo para formar el derivado 6'-hidroximetilo es catalizada por las enzimas CYP. CYP3A4 parece contribuir al metabolismo de etoricoxib in vivo. Los estudios in vitro indican que CYP2D6, CYP2C9, CYP1A2 y CYP2C19 también pueden catalizar la vía metabólica principal, pero no se han estudiado sus funciones cuantitativas in vivo. Cinco metabolitos han sido identificados en el hombre. El principal metabolito es el derivado del ácido 6'-carboxílico de etoricoxib formado por la oxidación adicional del derivado 6'-hidroximetilo. Estos metabolitos principales no muestran actividad medible o solo son débilmente activos como inhibidores de la COX-2. Ninguno de estos metabolitos inhibe la COX-1. Eliminación: Después de la administración de una única dosis intravenosa radiomarcada de 25 mg de etoricoxib a sujetos sanos, el 70% de la radioactividad se recuperó en la orina y el 20% en las heces, principalmente como metabolitos. Menos del 2% se recuperó como fármaco inalterado. La eliminación de etoricoxib ocurre casi exclusivamente a través del metabolismo seguido de la excreción renal. Las concentraciones de etoricoxib en el estado estacionario se alcanzan dentro de los 7 días de la administración de 120 mg 1 vez al día, con una tasa de acumulación de aproximadamente 2, que corresponde a una vida media de aproximadamente 22 horas. La eliminación plasmática después de una dosis intravenosa de 25 mg se estima en aproximadamente 50 ml/min. Características en pacientes: Pacientes de edad avanzada: la farmacocinética en los pacientes de edad avanzada (de 65 ańos o más) es similar a la de los jóvenes. Género: la farmacocinética de etoricoxib es similar entre hombres y mujeres. Insuficiencia hepática: los pacientes con disfunción hepática leve (puntuación de Child-Pugh de 5 a 6) que recibieron 60 mg de etoricoxib 1 vez al día tuvieron un AUC promedio aproximadamente 16% mayor en comparación con los sujetos sanos administrados con el mismo régimen. Los pacientes con disfunción hepática moderada (puntuación de Child-Pugh 7-9) a los que se les administró 60 mg de etoricoxib cada 2 días tuvieron una AUC media similar a los sujetos sanos que recibieron etoricoxib 60 mg 1 vez al día; etoricoxib 30 mg 1 vez al día no se ha estudiado en esta población. No hay datos clínicos o farmacocinéticos en pacientes con disfunción hepática grave (puntuación de Child-Pugh ≥10) (ver secciones Posología y Contraindicaciones) Pacientes pediátricos: no se ha estudiado la farmacocinética de etoricoxib en pacientes pediátricos (<12 ańos). En un estudio farmacocinético (n = 16) realizado en adolescentes (de 12 a 17 ańos), la farmacocinética en adolescentes con un peso de 40 a 60 kg, a los que se administró etoricoxib 60 mg 1 vez al día y adolescentes> 60 kg con etoricoxib 90 mg 1 vez al día fue similar a la farmacocinética en adultos que recibieron etoricoxib 90 mg 1 vez al día. No se han establecido la seguridad y la eficacia de etoricoxib en pacientes pediátricos (ver sección Posología). Datos de seguridad preclínicos: En estudios preclínicos, se ha demostrado que etoricoxib no es genotóxico. Etoricoxib no fue carcinogénico en ratones. Las ratas desarrollaron adenomas hepatocelulares y de células foliculares tiroideas a > 2 veces la dosis diaria en humanos [90 mg] en función de la exposición sistémica cuando se dosificaron diariamente durante aproximadamente 2 ańos. Los adenomas hepatocelulares y de células foliculares tiroideas observados en ratas se consideran una consecuencia del mecanismo específico de la rata relacionado con la inducción de la enzima CYP hepática. No se ha demostrado que etoricoxib cause la inducción de la enzima hepática CYP3A en humanos. En la rata, la toxicidad gastrointestinal de etoricoxib aumentó con la dosis y el tiempo de exposición. En el estudio de toxicidad de 14 semanas, etoricoxib causó úlceras gastrointestinales a exposiciones mayores que las observadas en el hombre a la dosis terapéutica. En el estudio de toxicidad de 53 y 106 semanas, también se observaron úlceras gastrointestinales con exposiciones comparables a las observadas en el hombre a la dosis terapéutica. En perros, se observaron anomalías renales y gastrointestinales a altas exposiciones. Etoricoxib no fue teratogénico en los estudios de toxicidad reproductiva realizados en ratas a 15 mg/kg/día (esto representa aproximadamente 1.5 veces la dosis diaria en humanos [90 mg] en función de la exposición sistémica). En conejos, se observó un aumento relacionado con el tratamiento de las malformaciones cardiovasculares a niveles de exposición por debajo de la exposición clínica a la dosis diaria en humanos (90 mg). Sin embargo, no se observaron malformaciones fetales ni esqueléticas externas relacionadas con el tratamiento. En ratas y conejos, hubo un aumento dependiente de la dosis en la pérdida post implantación a exposiciones mayores o iguales a 1.5 veces la exposición humana (ver secciones Contraindicaciones y Embarazo y lactancia).
Posología: Como los riesgos cardiovasculares de etoricoxib pueden aumentar con la dosis y la duración de la exposición, se debe utilizar la duración más corta y la dosis diaria efectiva más baja posible. La necesidad del paciente de alivio sintomático y respuesta al tratamiento deberá ser evaluada periódicamente, (ver secciones Contraindicaciones, Advertencias, Efectos colaterales y Propiedades farmacodinámicas). Artritis gotosa aguda: La dosis recomendada es de 120 mg 1 vez al día. Etoricoxib 120 mg solo debe ser empleada durante el período sintomático agudo, limitado a un máximo de 8 días de tratamiento. Analgesia: Dolor agudo y dismenorrea primaria: La dosificación recomendada es de 120 mg 1 vez al día. Etoricoxib 120 mg sólo debe emplearse durante el período sintomático agudo, limitado a un máximo de 8 días de tratamiento. Debido a que el riesgo cardiovascular de los inhibidores selectivos de COX-2 puede aumentar dependiendo de la dosis y el tiempo de exposición, se debe usar el menor tiempo posible y a la menor dosis efectiva diaria. Se debe re-evaluar la necesidad del paciente de alivio sintomático y respuesta a la terapia. Poblaciones especiales: Pacientes de edad avanzada: No es necesario ajustar la dosis en pacientes de edad avanzada. Al igual que con otros medicamentos, se debe tener precaución en pacientes de edad avanzada (ver sección Advertencias). Pacientes con insuficiencia hepática: Independientemente de la indicación, en pacientes con disfunción hepática leve (puntuación de Child-Pugh 5 - 6) no se debe exceder una dosis de 60 mg 1 vez al día. En pacientes con disfunción hepática moderada (puntuación de Child-Pugh 7 - 9), independientemente de la indicación, no debe excederse la dosis de 30 mg 1 vez al día. La experiencia clínica es limitada, particularmente en pacientes con disfunción hepática moderada y se recomienda precaución. No hay experiencia clínica en pacientes con disfunción hepática severa (puntuación de Child-Pugh ≥10); por lo tanto, su uso está contraindicado en estos pacientes (ver secciones Contraindicaciones, Advertencias y Propiedades farmacocinéticas). Pacientes con insuficiencia renal: No es necesario ajustar la dosis en pacientes con clearance de creatinina ≥30 ml/min (ver sección Propiedades farmacocinéticas). El uso de etoricoxib en pacientes con clearance de creatinina < 30 ml/min está contraindicado (ver secciones Contraindicaciones y Advertencias). Población pediátrica: No se han determinado la seguridad y la eficacia del etoricoxib en nińos.Etoricoxib está contraindicado en nińos y adolescentes menores de 16 ańos (ver sección Contraindicaciones). Método de administración: Etox se administra por vía oral y se puede tomar con o sin alimentos. El inicio del efecto del medicamento puede ser más rápido cuando Etox se administra sin alimentos. Esto debe considerarse cuando se necesita un alivio sintomático rápido.
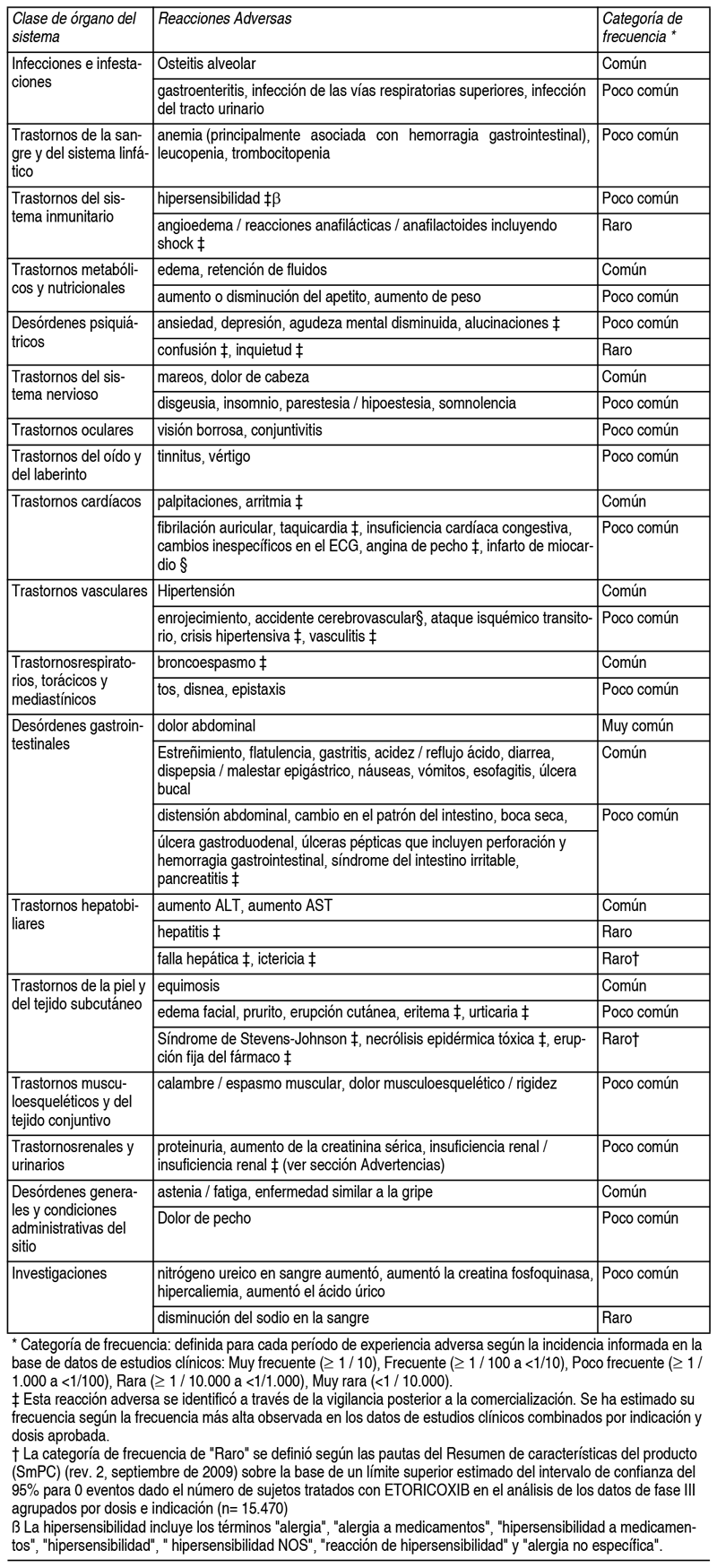
Efectos Colaterales:Resumen del perfil de seguridad: En estudios clínicos, se evaluó la seguridad de etoricoxib en 7.152 individuos, incluyendo 4.614 pacientes con OA, RA, dolor lumbar crónico o espondilitis anquilosante (aproximadamente 600 pacientes con artrosis o artritis reumatoide fueron tratados durante 1 ańo o más). En estudios clínicos, el perfil de efectos indeseables fue similar en pacientes con OA o RA tratados con etoricoxib durante 1 ańo o más. En un estudio clínico para la artritis gotosa aguda, los pacientes fueron tratados con etoricoxib 120 mg 1 vez al día durante 8 días. El perfil de experiencia adversa en este estudio fue en general similar al reportado en los estudios combinados de OA, RA y dolor lumbar crónico. En un programa de resultados de seguridad cardiovascular de datos combinados de 3 estudios controlados comparadores activos, 17.412 pacientes con OA o AR fueron tratados con etoricoxib (60 mg o 90 mg) durante una duración media de aproximadamente 18 meses. Los datos de seguridad y los detalles de este programa se presentan en la sección Propiedades farmacodinámicas. En estudios clínicos para dolor dental postoperatorio agudo después de cirugía que incluyó 614 pacientes tratados con etoricoxib (90 mg o 120 mg), el perfil de experiencia adversa en estos estudios fue generalmente similar al reportado en los estudios combinados de OA, RA y dolor lumbar crónico. Lista tabulada de reacciones adversas: Los siguientes efectos adversos se informaron con una incidencia mayor que el placebo en ensayos clínicos en pacientes con OA, RA, dolor lumbar crónico o espondilitis anquilosante tratados con etoricoxib 30 mg, 60 mg o 90 mg hasta la dosis recomendada durante hasta 12 semanas; en los estudios del programa MEDAL por hasta 3˝ ańos; en estudios de dolor agudo a corto plazo de hasta 7 días; o en la experiencia posterior a la comercialización (ver Tabla 1):
Los siguientes efectos adversos graves han sido reportados en asociación con el uso de AINEs y no se puede descartar la presencia de etoricoxib: nefrotoxicidad, incluida la nefritis intersticial y el síndrome nefrótico. Notificación de sospechas de reacciones adversas: Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del Subdepartamento de Farmacovigilancia de la Agencia Nacional de Medicamentos: http://www.ispch.cl/sites/default/files/Instructivo%20para%20completar%20formulario%20RAM_0.p df.
Contraindicaciones:Hipersensibilidad: debido a la potencial hipersensibilidad cruzada con otros AINEs, no debe administrarse a pacientes que han sufrido síntomas de asma, rinitis, urticaria, pólipos nasales, angioedema, broncoespasmo y otros síntomas o reacciones alérgicas o anafilactoideas asociadas a ácido acetilsalicílico u otro AINE. En raros casos se han presentado reacciones anafilácticas fatales y asmáticas severas. No debe usarse AINEs con excepción de ácido acetilsalicílico en pacientes en el período post operatorio inmediato a una cirugía de by-pass coronario. Hipersensibilidad a la sustancia activa o a cualquiera de los excipientes enumerados en la sección Excipientes: Ulcera péptica activa o hemorragia gastrointestinal (GI) activa. Embarazo y lactancia (ver secciones Embarazo y lactancia y Datos de seguridad preclínica). Disfunción hepática severa (albúmina sérica <25 g/l o puntuación Child-Pugh ≥10). Clearance estimado de creatinina renal <30 ml/min. Nińos y adolescentes menores de 16 ańos. Enfermedad inflamatoria intestinal. Insuficiencia cardíaca congestiva (NYHA II-IV). Pacientes con hipertensión cuya presión arterial está persistentemente elevada por encima de 140/90 mm Hg y no ha sido controlada adecuadamente. El uso de estos medicamentos está contraindicado en pacientes que padezcan enfermedad isquémica cardíaca, hayan presentado un accidente isquémico cerebral o tengan insuficiencia cardíaca de grado II a IV. Hipersensibilidad a sulfonamidas u otros AINEs.
Advertencias:Advertencias y precauciones especiales de uso: Efectos gastrointestinales: Síntomas de toxicidad gastrointestinal severa tales como inflamación, sangramiento, ulceración y perforación del intestino grueso y delgado pueden ocurrir en cualquier momento con o sin síntomas previos, en pacientes en terapia crónica con AINEs, por lo que se debe estar alerta frente a la presencia de síntomas de ulceración o sangrado. Se recomienda precaución en el tratamiento de pacientes con mayor riesgo de desarrollar una complicación gastrointestinal con AINES; ancianos, pacientes que usan cualquier otro AINE o ácido acetilsalicílico concomitante o pacientes con antecedentes de enfermedad gastrointestinal, como ulceración y hemorragia digestiva. Efectuar seguimiento de los pacientes en tratamiento crónico con AINEs por signos o síntomas de ulceración o sangramiento gastrointestinal. Existe el riesgo de sangramiento estomacal severo, que potencialmente amenaza la vida. El uso concomitante con ácido acetilsalicílico incluso a dosis bajas aumenta el riesgo de úlcera gastrointestinal y sus complicaciones. Efectos cardiovasculares: Al prescribir estos fármacos, los médicos deben tener especial precaución si los pacientes presentan factores de riesgo cardiovascular como hipertensión arterial, hiperlipidemia, diabetes mellitus, si son fumadores o presentan enfermedad arterial periférica. Se debe utilizar la dosis mínima efectiva con la que se obtengan efectos beneficiosos y la duración del tratamiento debe ser lo más corta posible. La necesidad de continuar el tratamiento debe ser evaluada periódicamente. Debido al riesgo de que se produzcan eventos cardiovasculares severos con el uso de AINEs, a excepción del ácido acetilsalicílico, debe evaluarse cuidadosamente la condición del paciente antes de prescribir estos medicamentos. Usar con precaución en pacientes con compromiso de la función cardíaca, hipertensión, terapia diurética crónica y otras condiciones que predisponen a retención de fluidos, debido a que los AINEs pueden causar la retención de fluidos además de edema periférico. Estudios clínicos sugieren que los inhibidores selectivos de la COX-2 pueden estar asociados con un riesgo de eventos trombóticos (especialmente infarto de miocardio [IM] y accidente cerebrovascular), en relación con el placebo y algunos AINE. Como los riesgos cardiovasculares de etoricoxib pueden aumentar con la dosis y la duración de la exposición, se debe utilizar la dosis más corta posible efectiva y la dosis diaria efectiva más baja. La necesidad del paciente de alivio sintomático y respuesta al tratamiento se debe volver a evaluar periódicamente, especialmente en pacientes con osteoartritis (ver secciones Posología, Contraindicaciones, Efectos colaterales y Propiedades farmacodinámicas). Los pacientes con factores de riesgo significativos de eventos cardiovasculares (por ejemplo, hipertensión, hiperlipidemia, diabetes mellitus, tabaquismo) solo deben tratarse con etoricoxib después de una consideración cuidadosa (ver sección Propiedades farmacodinámicas). Los inhibidores selectivos de COX-2 no son un sustituto del ácido acetilsalicílico para la profilaxis de enfermedades tromboembólicas cardiovasculares debido a su falta de efecto antiplaquetario. Por lo tanto, las terapias antiplaquetarias no deben suspenderse (ver las secciones anteriores, Interacciones y Propiedades farmacodinámicas). Efectos renales: Se puede producir insuficiencia renal aguda, nefritis intersticial con hematuria, síndrome nefrótico, proteinuria, hiperkalemia, hiponatremia, necrosis papilar renal y otros cambios medulares renales. Pacientes con falla renal preexistente están con mayor riesgo de sufrir insuficiencia renal aguda. Una descompensación renal se puede precipitar en pacientes en tratamiento por AINEs, debido a una reducción dosis dependiente en la formación de prostaglandinas afectando principalmente a ancianos, lactantes, prematuros, pacientes con falla renal, cardíaca o disfunción hepática, glomerulonefritis crónica, deshidratación, diabetes mellitus, septicemia, pielonefritis y depleción de volumen extracelular en aquellos que están tomando inhibidores de la ECA, y/o diuréticos. Las prostaglandinas renales pueden jugar un papel compensador en el mantenimiento de la perfusión renal. Por lo tanto, bajo condiciones de perfusión renal comprometida, la administración de etoricoxib puede causar una reducción en la formación de prostaglandinas y, secundariamente, en el flujo sanguíneo renal, y por lo tanto deteriorar la función renal. Los pacientes con mayor riesgo de esta respuesta son aquellos con una función renal significativamente preexistente, insuficiencia cardíaca descompensada o cirrosis. Se debe considerar la monitorización de la función renal en tales pacientes. Retención de líquido, edema e hipertensión: Al igual que con otros medicamentos conocidos por inhibir la síntesis de prostaglandinas, se ha observado retención de líquido, edema e hipertensión en pacientes que toman etoricoxib. Todos los medicamentos antiinflamatorios no esteroideos (AINES), incluido el etoricoxib, pueden estar asociados con un nuevo inicio o insuficiencia cardíaca congestiva recurrente. Para obtener información sobre la respuesta relacionada con la dosis de etoricoxib, ver la sección Propiedades farmacodinámicas. Se debe tener precaución en pacientes con antecedentes de insuficiencia cardíaca, disfunción ventricular izquierda o hipertensión y en pacientes con edema preexistente por cualquier otro motivo. Si hay evidencia clínica de deterioro en la condición de estos pacientes, se deben tomar las medidas apropiadas, incluida la interrupción del tratamiento con etoricoxib. Etoricoxib puede estar asociado con hipertensión más frecuente y severa que algunos otros AINE e inhibidores selectivos de la COX-2, particularmente a dosis altas. Por lo tanto, la hipertensión debe controlarse antes del tratamiento con etoricoxib (ver sección Contraindicaciones) y se debe prestar especial atención al control de la presión arterial durante el tratamiento con etoricoxib. La presión arterial debe controlarse dentro de las 2 semanas posteriores al inicio del tratamiento y periódicamente a partir de ese momento. Si la presión arterial aumenta significativamente, se debe considerar un tratamiento alternativo. Efectos hepáticos: Se han notificado elevaciones de alanina aminotransferasa (ALT) y/o aspartato aminotransferasa (AST) (aproximadamente 3 o más veces el límite superior de lo normal) en aproximadamente 1% de los pacientes de estudios clínicos tratados hasta 1 ańo con etoricoxib 30, 60 y 90 mg diarios. Debe controlarse a todos los pacientes con síntomas y/o signos que sugieran disfunción hepática o en los que se haya producido una prueba de función hepática anormal. Si se presentan signos de insuficiencia hepática o si se detectan pruebas de función hepática persistentemente anormales (3 veces el límite superior de lo normal), debe suspenderse etoricoxib. Efectuar monitoreo de transaminasas y enzimas hepáticas en pacientes en tratamiento con AINEs, especialmente en aquellos tratados con nimesulida, sulindaco, diclofenaco y naproxeno. General: Se han producido reacciones anafilactoideas en pacientes asmáticos, sin exposición previa a AINEs, pero que han experimentado previamente rinitis con o sin pólipos nasales o que exhiben broncoespasmo potencialmente fatal después de tomar ácido acetilsalicílico u otro AINE. Si durante el tratamiento, los pacientes sufren de un deterioro en cualquiera de las funciones del sistema orgánico descritas anteriormente, se deben tomar las medidas apropiadas y se debe considerar la interrupción del tratamiento con etoricoxib. Se debe mantener una supervisión médicamente apropiada cuando se usa etoricoxib en ancianos y en pacientes con disfunción renal, hepática o cardíaca. Se debe tener precaución al iniciar el tratamiento con etoricoxib en pacientes con deshidratación. Es aconsejable rehidratar a los pacientes antes de comenzar la terapia con etoricoxib. Se han notificado muy raramente, reacciones cutáneas graves, algunas de ellas mortales, que incluyen dermatitis exfoliativa, síndrome de Stevens-Johnson y necrólisis epidérmica tóxica, en pacientes en tratamiento con valdecoxib; por lo que se debe discontinuar el medicamento ante la aparición de rash cutáneo o ante los primeros signos de hipersensibilidad. Se han notificado reacciones de hipersensibilidad graves (como anafilaxia y angioedema) en pacientes que reciben etoricoxib (ver sección Efectos colaterales). Algunos inhibidores selectivos de la COX-2 se han asociado con un mayor riesgo de reacciones cutáneas en pacientes con antecedentes de alergia a algún medicamento. Etoricoxib debe suspenderse a la primera aparición de erupción cutánea, lesiones en la mucosa o cualquier otro signo de hipersensibilidad. Etoricoxib puede enmascarar la fiebre y otros signos de inflamación. Se debe tener precaución cuando se administre etoricoxib conjuntamente con warfarina u otros anticoagulantes orales (ver sección Interacciones). El uso de etoricoxib, así como con cualquier medicamento que se sabe que inhibe la síntesis de ciclooxigenasa / prostaglandina, no se recomienda en mujeres que intentan concebir (ver secciones Embarazo y lactancia, Propiedades farmacodinámicas y Datos de seguridad preclínica). Los comprimidos de Etox contienen lactosa. Los pacientes con intolerancia hereditaria a galactosa, insuficiencia de lactasa de Lapp o malabsorción de glucosa o galactosa no deben tomar este medicamento.
Precauciones:Fertilidad, embarazo y lactancia: Embarazo: No hay datos clínicos sobre embarazos expuestos disponibles para etoricoxib. Los estudios en animales han mostrado toxicidad reproductiva (ver sección Datos de seguridad preclínica). El potencial de riesgo humano en el embarazo es desconocido. Etoricoxib, como ocurre con otros medicamentos que inhiben la síntesis de prostaglandinas, puede causar inercia uterina y el cierre prematuro del ductus arterioso durante el último trimestre. Etoricoxib está contraindicado durante el embarazo (ver sección Contraindicaciones). Si una mujer queda embarazada durante el tratamiento, etoricoxib debe suspenderse. Lactancia: No se sabe si etoricoxib se excreta en la leche humana. Etoricoxib se excreta en la leche de ratas lactantes. Las mujeres que usan etoricoxib no deben amamantar (ver secciones Contraindicaciones y Datos de seguridad preclínica). Fertilidad: El uso de etoricoxib, así como cualquier otra sustancia farmacológica que se sabe que inhibe la COX-2, no se recomienda en mujeres que intentan concebir. Efectos sobre la capacidad para conducir y utilizar máquinas: Los pacientes que experimenten mareos, vértigo o somnolencia mientras toman etoricoxib deben abstenerse de conducir o manejar maquinaria.
Interacciones Medicamentosas:Interacciones farmacodinámicas: Anticoagulantes orales: en sujetos estabilizados con warfarina crónica, la administración de etoricoxib 120 mg al día se asoció con un aumento aproximado del 13% en el tiempo de protrombina International Normalized Ratio (INR). Por lo tanto, los pacientes que reciben anticoagulantes orales deben ser estrechamente monitorizados por su tiempo de protrombina INR, particularmente en los primeros días cuando se inicia el tratamiento con etoricoxib o cuando se cambia la dosis de etoricoxib (ver sección Advertencias). Diuréticos, inhibidores de la ECA y antagonistas de la angiotensina II: los AINEs pueden reducir el efecto de los diuréticos y otros fármacos antihipertensivos. En algunos pacientes con función renal comprometida (Ej. Pacientes deshidratados o ancianos con función renal comprometida) la administración concomitante de un inhibidor de la ECA o antagonista de la angiotensina II y agentes que inhiben la ciclooxigenasa puede provocar un mayor deterioro de la función renal, incluida una posible insuficiencia renal, que generalmente es reversible. Estas interacciones deben considerarse en pacientes que toman etoricoxib concomitantemente con inhibidores de la ECA o antagonistas de la angiotensina II. Por lo tanto, la combinación debe administrarse con precaución, especialmente en los ancianos. Los pacientes deben estar adecuadamente hidratados y se debe considerar la monitorización de la función renal después del inicio de la terapia concomitante, y periódicamente a partir de entonces. Ácido acetilsalicílico: en un estudio en sujetos sanos, en estado de equilibrio, etoricoxib 120 mg 1 vez al día no tuvo efecto sobre la actividad antiplaquetaria del ácido acetilsalicílico (81 mg 1 vez al día). Etoricoxib se puede usar concomitantemente con ácido acetilsalicílico en dosis utilizadas para la profilaxis cardiovascular (ácido acetilsalicílico en dosis bajas). Sin embargo, la administración concomitante de dosis bajas de ácido acetilsalicílico con etoricoxib puede dar como resultado un aumento en la tasa de ulceración gastrointestinal u otras complicaciones en comparación con el uso de etoricoxib solo. No se recomienda la administración concomitante de etoricoxib con dosis de ácido acetilsalicílico superiores a las de la profilaxis cardiovascular o con otros AINE (ver secciones Propiedades farmacodinámicas y Advertencias). Ciclosporina y tacrolimus: aunque esta interacción no se ha estudiado con etoricoxib, la administración concomitante de ciclosporina o tacrolimus con cualquier AINE puede aumentar el efecto nefrotóxico de ciclosporina o tacrolimus. La función renal debe controlarse cuando etoricoxib y cualquiera de estos medicamentos se usa en combinación. Interacciones farmacocinéticas: El efecto de etoricoxib sobre la farmacocinética de otras drogas: Litio: AINEs reducen la excreción renal de litio y, por lo tanto, aumentan los niveles plasmáticos de litio. Si es necesario, controle de cerca el litio sanguíneo y ajuste la dosis de litio mientras se toma la combinación y cuando se retira el AINE. Metotrexato: 2 estudios investigaron los efectos de etoricoxib 60, 90 ó 120 mg administrados 1 vez al día durante 7 días en pacientes que recibieron dosis de metotrexato 1 vez a la semana de 7.5 a 20 mg para la artritis reumatoide. Etoricoxib a 60 y 90 mg no tuvo efecto sobre las concentraciones plasmáticas de metotrexato o el clearance renal. En un estudio, etoricoxib 120 mg no tuvo efecto, pero en el otro estudio, etoricoxib 120 mg aumentó las concentraciones plasmáticas de metotrexato en un 28% y redujo el clearance renal de metotrexato en un 13%. Se recomienda una monitorización adecuada de la toxicidad relacionada con el metotrexato cuando se administren etoricoxib y metotrexato de forma concomitante. Anticonceptivos orales: Etoricoxib 60 mg administrado concomitantemente con un anticonceptivo oral que contiene 35 mcg de etinilestradiol (EE) y 0.5 a 1 mg de noretindrona durante 21 días aumentó el AUC0-24hr al estado estacionario de EE en un 37%. Etoricoxib 120 mg administrado con el mismo anticonceptivo oral de manera concomitante o separada por 12 horas, aumentó la AUC0-24hr al estado estacionario de la EA en un 50 a 60%. Este aumento en la concentración de EE debe ser considerado al seleccionar un anticonceptivo oral para usar con etoricoxib. Un aumento en la exposición a EE puede aumentar la incidencia de eventos adversos asociados con anticonceptivos orales (Ej., Eventos tromboembólicos venosos en mujeres en riesgo). Terapia de reemplazo hormonal (HRT): Administración de 120 mg de etoricoxib con terapia de reemplazo hormonal consistente en estrógenos conjugados (0.625 mg PrematinTM) durante 28 días, aumentó el promedio de la AUC0-24hr en estado estacionario de la estrona no conjugada (41%), equilin (76%) y 17-β-estradiol (22%). No se ha estudiado el efecto de las dosis crónicas recomendadas de etoricoxib (30, 60 y 90 mg). Los efectos de etoricoxib 120 mg sobre la exposición (AUC0-24hr) a estos componentes estrogénicos de Premarin fueron menos de la mitad de los observados cuando se administró Premarin solo y la dosis aumentó de 0.625 a 1.25 mg. Se desconoce la importancia clínica de estos aumentos, y no se estudiaron dosis más altas de Premarin en combinación con etoricoxib. Estos aumentos en la concentración estrogénica deben tenerse en cuenta al seleccionar la terapia hormonal posmenopáusica para su uso con etoricoxib, ya que el aumento en la exposición a estrógenos puede aumentar el riesgo de eventos adversos asociados con la TRH. Prednisona / prednisolona: en estudios de interacción con medicamentos, etoricoxib no tuvo efectos clínicamente importantes sobre la farmacocinética de la prednisona / prednisolona. Digoxina: Etoricoxib 120 mg administrados 1 vez al día durante 10 días a voluntarios sanos no alteró el AUC0-24hr plasmático en estado estacionario ni la eliminación renal de digoxina. Hubo un aumento en la Cmáx de digoxina (aproximadamente 33%). Este aumento generalmente no es importante para la mayoría de los pacientes. Sin embargo, los pacientes con alto riesgo de toxicidad por digoxina deben ser monitoreados cuando se administra etoricoxib y digoxina de forma concomitante. Efecto de etoricoxib en fármacos metabolizados por sulfotransferasas: Etoricoxib es un inhibidor de la actividad de la sulfotransferasa humana, particularmente SULT1E1, y se ha demostrado que aumenta las concentraciones séricas de etinilestradiol. Si bien el conocimiento sobre los efectos de las sulfotransferasas múltiples es actualmente limitado y las consecuencias clínicas para muchos fármacos aún se están examinando, puede ser prudente tener cuidado cuando se administra etoricoxib simultáneamente con otros fármacos metabolizados principalmente por las sulfotransferasas humanas (por ejemplo, salbutamol oral y minoxidil). Efecto de etoricoxib en fármacos metabolizados por isoenzimas CYP: Según estudios in vitro, no se espera que etoricoxib inhiba los citocromos P450 (CYP) 1A2, 2C9, 2C19, 2D6, 2E1 o 3A4. En un estudio en sujetos sanos, la administración diaria de 120 mg de etoricoxib no alteró la actividad hepática de CYP3A4 evaluada mediante la prueba de aliento con eritromicina. Efectos de otros medicamentos en la farmacocinética de etoricoxib: La vía principal del metabolismo de etoricoxib depende de las enzimas CYP. CYP3A4 parece contribuir al metabolismo de etoricoxib in vivo. Los estudios in vitro indican que CYP2D6, CYP2C9, CYP1A2 y CYP2C19 también pueden catalizar la ruta metabólica principal, pero sus roles cuantitativos no se han estudiado in vivo. Ketoconazol: Ketoconazol, un potente inhibidor de CYP3A4, dosificado a 400 mg 1 vez al día durante 11 días a voluntarios sanos, no tuvo ningún efecto clínicamente importante en la farmacocinética de dosis única de 60 mg de etoricoxib (43% de aumento en el AUC). Voriconazol y miconazol: la administración concomitante de voriconazol oral o gel oral tópico de miconazol, inhibidores potentes de CYP3A4, con etoricoxib causó un ligero aumento de la exposición a etoricoxib, pero no se considera clínicamente significativo según los datos publicados. Rifampicina: la administración concomitante de etoricoxib con rifampicina, un potente inductor de las enzimas CYP, produjo una disminución del 65% en las concentraciones plasmáticas de etoricoxib. Esta interacción puede provocar la recurrencia de los síntomas cuando etoricoxib se coadministra con rifampicina. Si bien esta información puede sugerir un aumento en la dosis, las dosis de etoricoxib mayores que las enumeradas para cada indicación no se han estudiado en combinación con rifampicina y, por lo tanto, no se recomiendan (ver sección Posología). Antiácidos: los antiácidos no afectan la farmacocinética de etoricoxib en un grado clínicamente relevante.
Sobredosificación: En estudios clínicos, la administración de dosis únicas de etoricoxib hasta 500 mg y dosis múltiples hasta 150 mg/día durante 21 días no produjo toxicidad significativa. Se han notificado casos de sobredosis aguda con etoricoxib, aunque no se informaron experiencias adversas en la mayoría de los casos. Las experiencias adversas observadas con mayor frecuencia fueron consistentes con el perfil de seguridad para etoricoxib (por ejemplo, eventos gastrointestinales, eventos cardiorenales). En caso de sobredosis, es razonable emplear las medidas de soporte usuales, por ejemplo, eliminar el material no absorbido del tracto gastrointestinal, emplear la monitorización clínica e instituir terapia de apoyo, si es necesario. Etoricoxib no es dializable por hemodiálisis; no se sabe si etoricoxib es dializable por diálisis peritoneal.
Incompatibilidades: No aplica.
Conservación:Precauciones especiales de almacenamiento: Este medicamento no requiere ninguna condición especial de almacenamiento.
Presentaciones: Estuche conteniendo blíster PA/aluminio/PVC-aluminio con X comprimidos recubiertos.
 Laboratorio
Laboratorio