Composición: Belsomra está disponible en comprimidos recubiertos que contienen 10 mg de Suvorexant. Ingrediente activo: Suvorexant. Cada comprimido recubierto contiene los siguientes ingredientes inactivos: Copovidona, Lactosa Monohidrato, Celulosa Microcristalina, Croscarmelosa Sódica y Estearato de Magnesio. Además, el recubrimiento contiene los siguientes ingredientes inactivos: Lactosa Monohidrato, Hipromelosa, Dióxido de Titanio y Triacetina. El recubrimiento para los comprimidos de 10 mg contiene también Oxido de Hierro Amarillo, Colorante FD&C N° 1, Laca Alumínica.
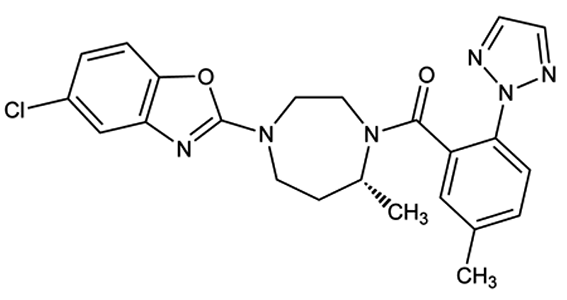
Descripción:Química: Suvorexant se describe químicamente como: [(7R)-4-(5-cloro-2-benzoxazolil)hexahidro-7-metil-1H-1,4-diazepin-1-il][5-metil-2-(2H-1,2,3-triazol-2-il)fenil]metanona. Su fórmula empírica es C23H23ClN6O2 y el peso molecular es 450,92. Su fórmula estructural es:
Suvorexant es un polvo de color blanco a blanquecino, insoluble en agua.
Indicaciones: Belsomra® está indicado para el tratamiento del insomnio, caracterizado por dificultades con la aparición del sueńo y/o la mantención del sueńo.
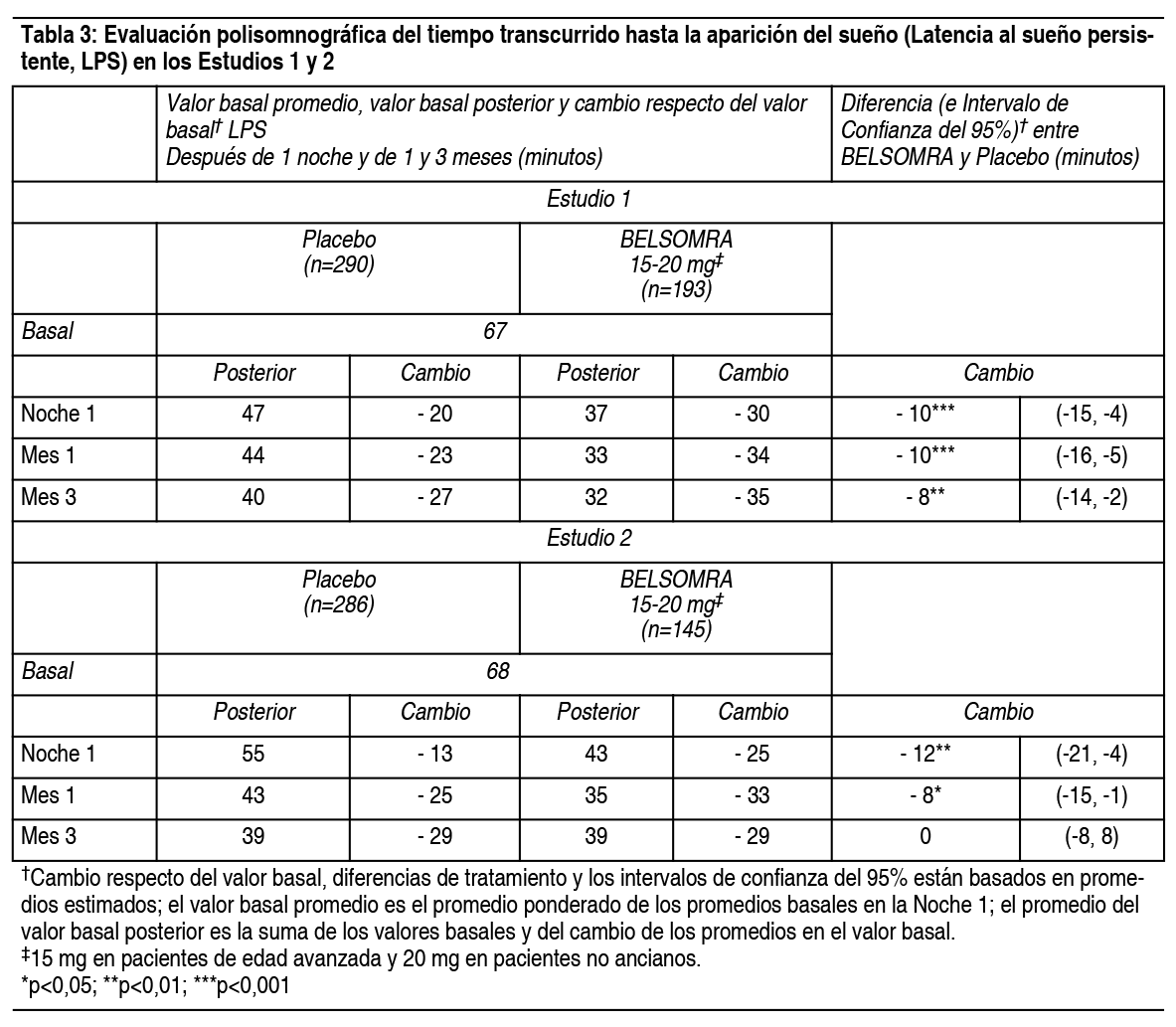
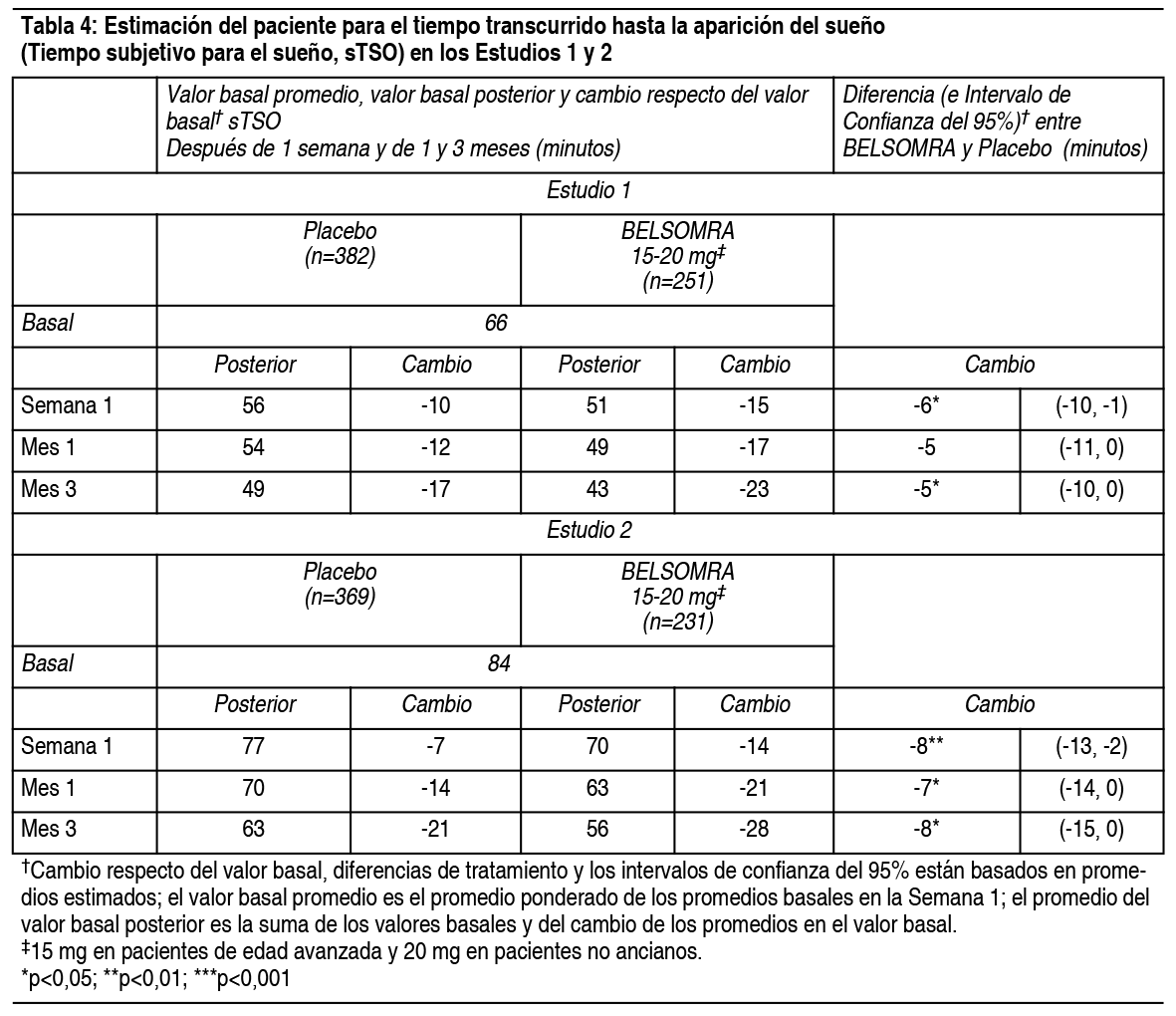
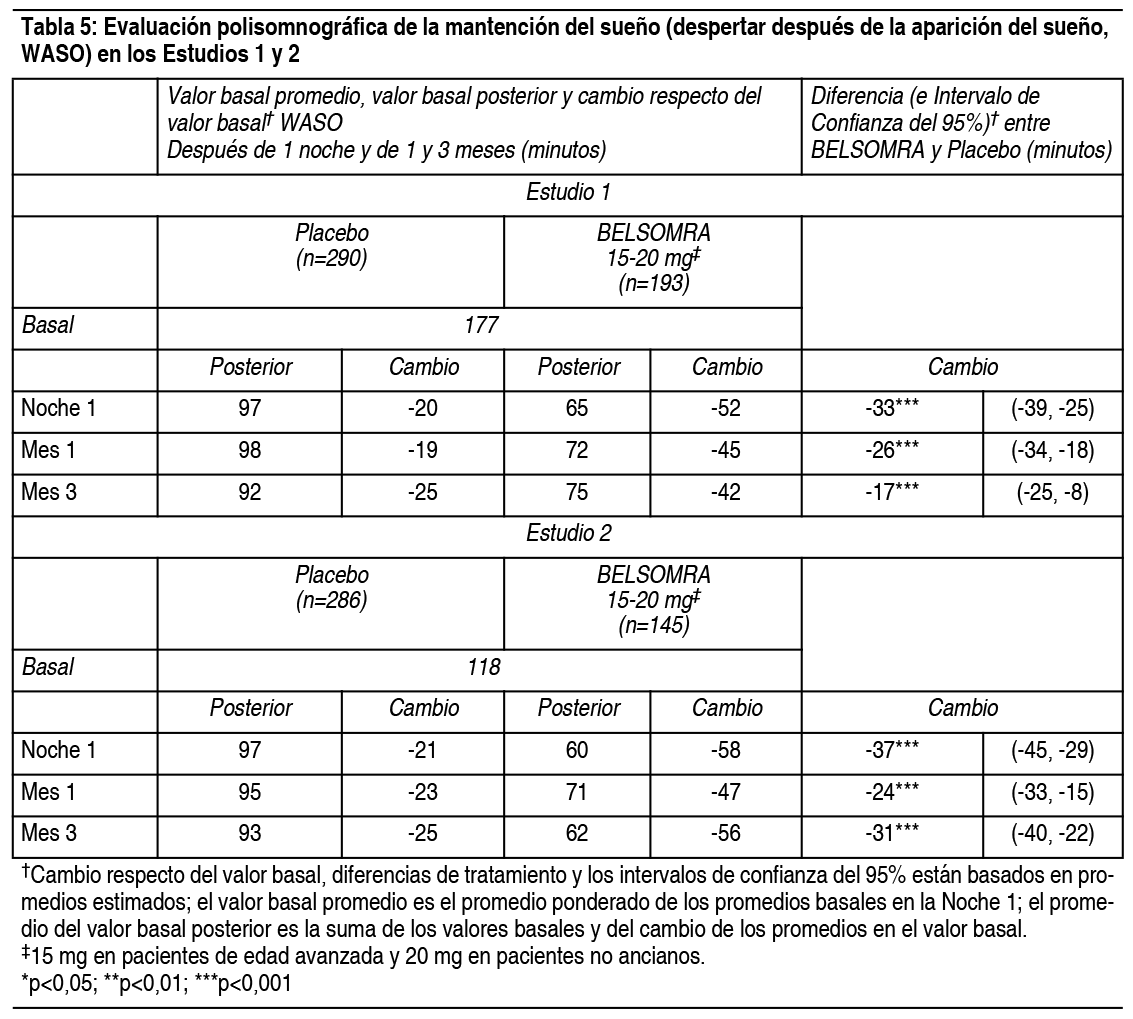
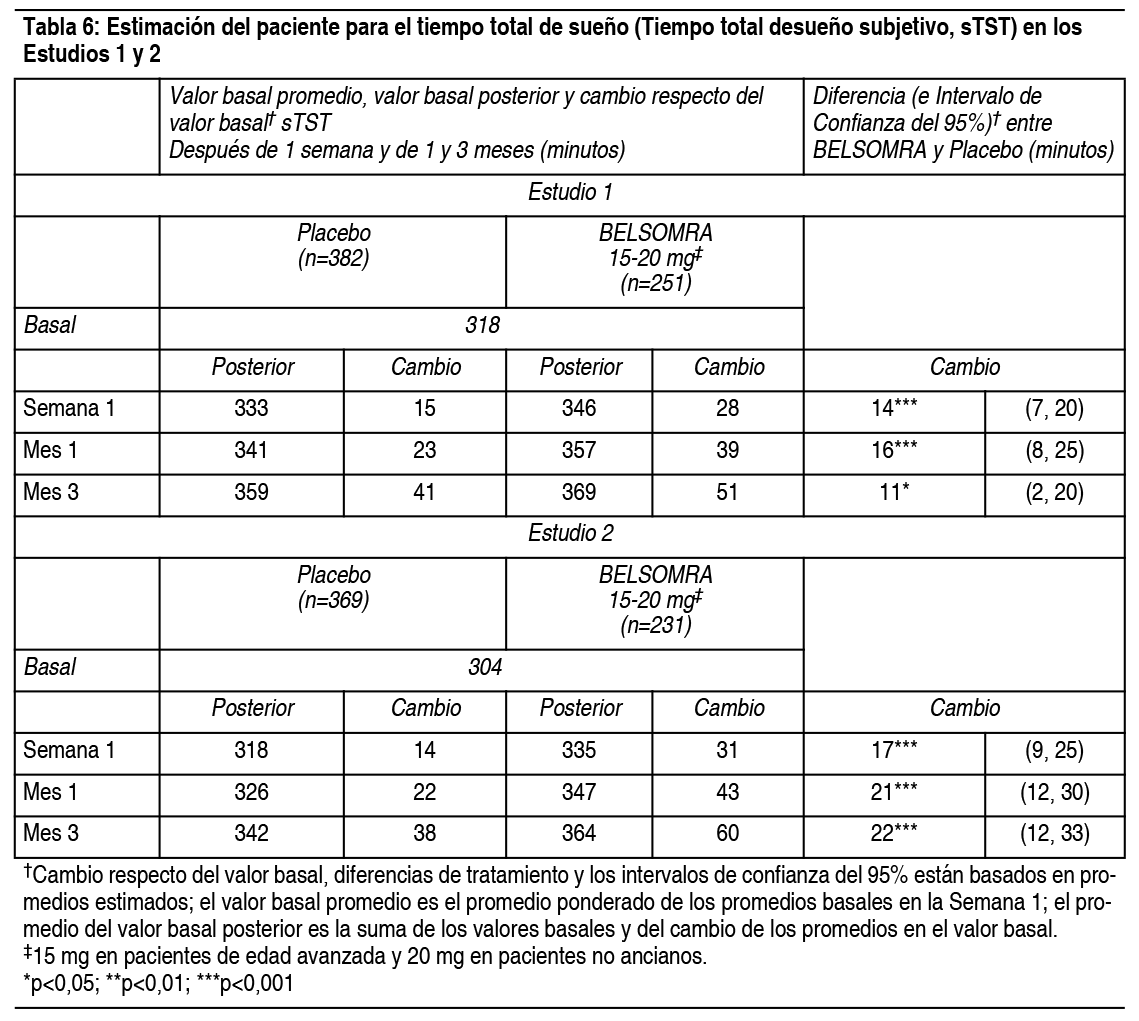
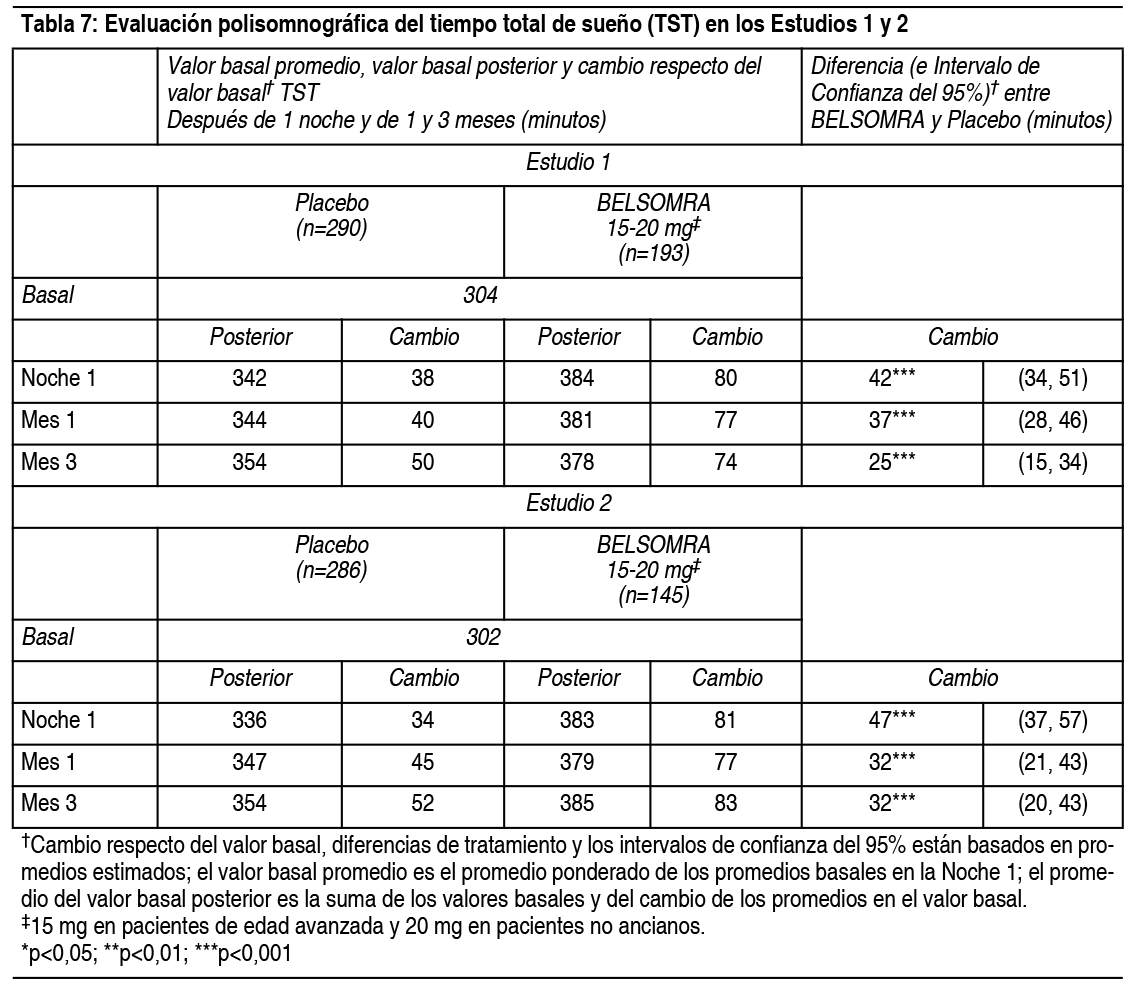
Propiedades:Estudios clínicos controlados: Estudios pivotales de eficacia: Se evaluó Belsomra en 2 estudios de diseńo similar, 3 meses de duración, aleatorizados, doble ciego, controlados con placebo, con grupos paralelos (Estudio 1 y Estudio 2) en pacientes con insomnio caracterizado por dificultades con la aparición del sueńo y la mantención del sueńo. En ambos estudios, los pacientes no ancianos (18-64 ańos) y de edad avanzada (≥65 ańos) fueron aleatorizados de manera separada. Tomando los estudios juntos, los adultos no ancianos (edad promedio 46 ańos; 465 mujeres, 275 hombres) fueron tratados con 20 mg de Belsomra (n=291) o placebo (n=449). Los pacientes de edad avanzada (edad promedio 71 ańos; 3346 mujeres, 174 hombres) fueron tratados con 15 mg de Belsomra (n=202) o placebo (n=318). En estos estudios, Belsomra en dosis de 15 mg o 20 mg resultó superior a placebo en la latencia al sueńo, de acuerdo con la evaluación objetiva mediante polisomnografía (Tabla 3) como también con la evaluación subjetiva de la latencia del sueńo estimada por el paciente (Tabla 4). Belsomra en dosis de 15 mg o 20 mg también fue superior a placebo en la mantención del sueńo, tal como lo indica la evaluación objetiva mediante polisomnografía (Tabla 5) como también con la evaluación subjetiva de la mantención del sueńo estimada por el paciente (Tabla 6). Análisis adicionales demostraron que el tratamiento con Belsomra aumentó el tiempo total de sueńo objetivo durante toda la noche (Tabla 7) y disminuyó el tiempo objetivo que pasó despierto después de la primera hora durante la Noche 1 (Tabla 8). La eficacia de Belsomra resultó similar entre mujeres y hombres y, basado en datos limitados, entre sujetos de raza blanca y no blanca. Un 27% de los pacientes tratados con 15 mg o 20 mg de Belsomra en el Estudio 1 y Estudio 2 no eran de raza blanca. La mayoría de los pacientes (69%) que no eran de raza blanca eran asiáticos.
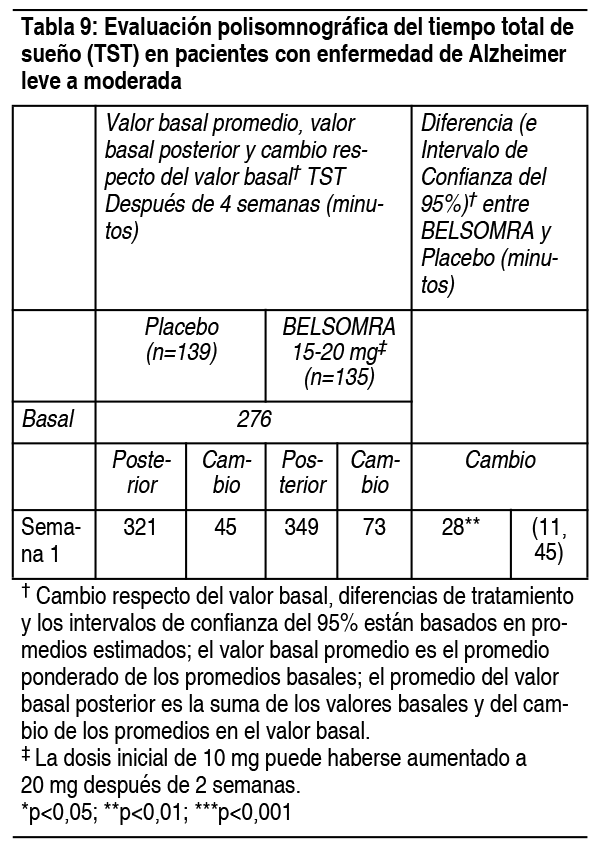
También se evaluó Belsomra en dosis de 30 mg y 40 mg en los estudios controlados con placebo, de 3 meses de duración (Estudio 1 y Estudio 2). Se observó que las dosis más altas tenían una eficacia similar que las dosis más bajas, pero se reportaron más reacciones adversas con las dosis mayores. Estudio de eficacia en pacientes con enfermedad de Alzheimer leve a moderada: Belsomra se evaluó en pacientes con enfermedad de Alzheimer de leve a moderada e insomnio caracterizado por dificultades con el inicio del sueńo y el mantenimiento del sueńo. Se realizó un estudio aleatorizado, doble ciego, controlado con placebo, de grupos paralelos, de multi-sitio de 4 semanas de Belsomra en pacientes con enfermedad de Alzheimer de leve a moderada (n=285) para el tratamiento del insomnio. Hombres y mujeres de 50-90 ańos (inclusive) de edad fueron tratados con Belsomra (n=142) o placebo (n=143). Los pacientes tratados con Belsomra recibieron 10 mg durante aproximadamente 14 días, de los cuales 76.8% aumentaron a 20 mg durante aproximadamente 14 días adicionales. En este estudio, Belsomra fue superior al placebo para el Tiempo Total de Sueńo (TST, por sus siglas es inglés) y el Mantenimiento del Sueńo (Wakefulness After Sleep Onset: WASO), según lo evaluó objetivamente la polisomnografía en la semana 4. Los pacientes tratados con Belsomra mostraron una mejoría estadísticamente significativa para ambos TST (p=0.001) (Tabla 9) y WASO (p=0.014) (Tabla 10), en comparación con los tratados con placebo.
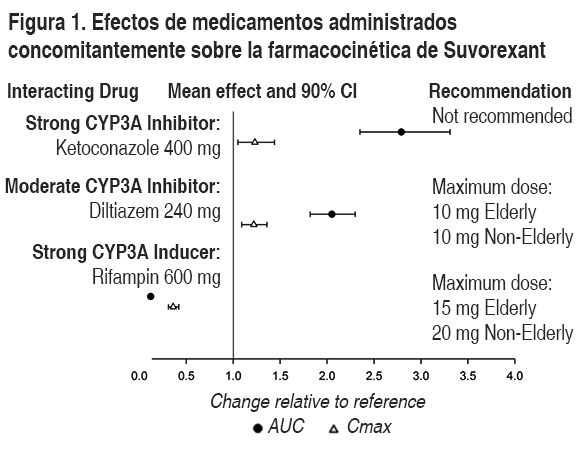
Estudios especiales sobre la seguridad: Efectos residuales al día siguiente:Estudios sobre la conducción: 2 estudios cruzados, de 4 períodos, aleatorizados, doble ciego, controlados con placebo y agente activo, evaluaron los efectos de la administración en la noche de Belsomra sobre el desempeńo al conducir la mańana siguiente, 9 horas después de la administración, en 24 sujetos sanos de edad avanzada (≥65 ańos, edad promedio 69 ańos; 14 hombres, 10 mujeres) y 28 sujetos no ancianos (edad promedio 46 ańos; 13 hombres, 15 mujeres). El criterio principal de valorcación fue el cambio en la desviación estándar promedio de la posición en el carril (SDLP, por sus siglas en inglés), una medida del desempeńo al conducir. No se observó una alteración significativa del desempeńo al conducir al día siguiente, según la evaluación del SDLP después de 1 noche y 8 noches seguidas de administración de suvorexant en dosis de 15 ó 30 mg en sujetos de edad avanzada, y 20 ó 40 mg en sujetos no ancianos. Sin embargo, algunos sujetos presentaron cambios en el SDLP indicativos de un deterioro en el desempeńo al conducir luego del tratamiento con suvorexant. En estos 2 estudios hubo 5 sujetos (4 mujeres no ancianas que recibieron suvorexant; 1 mujer de edad avanzada que recibió placebo) que suspendieron prematuramente sus pruebas de condución debido a somnolencia. Debido a las variaciones individuales, se debe aconsejar a los pacientes respecto de no conducir, operar maquinaria ni involucrarse en otras actividades que requieran una comple alerta mental hasta que se encuentren totalmente despiertos (ver Advertencias y Precauciones). Efectos sobre la memoria y el equilibrio al día siguiente en ancianos y no ancianos: 4 estudios controlados con placebo evaluaron los efectos de la administración en la noche de Belsomra sobre la memoria y el equilibrio al día siguiente, usando pruebas de aprendizaje de palabras y pruebas de balanceo corporal, respectivamente. 3 estudios no mostraron efectos significativos sobre la memoria o el equilibrio comparado con placebo. En un 4° estudio realizado en sujetos sanos no ancianos se observó una disminución significativa en el recuerdo de palabras después que se presentaran palabras a sujetos en la mańana luego de una dosis única de 40 mg de suvorexant, y se observó un aumento significativo en el área de balanceo corporal en la mańana luego de una dosis única de 20 ó 40 mg de suvorexant. Seguridad a mitad de la noche en sujetos de edad avanzada: Un estudio doble ciego, aleatorizado, controlado con placebo, en sujetos sanos de edad avanzada (n=12) evaluó el efecto de una dosis única de Belsomra sobre el equilibrio, la memoria y el desempeńo psicomotor después de ser despertado durante la noche. La administración en la noche de 30 mg de Belsomra no resultó en una alteración del equilibrio (medido por intermedio del área de balanceo corporal) o del desempeńo psicomotor (medido por el tiempo de reacción a la elección) a las 4 y 8 horas posteriores a la dosis comparado con placebo. Se observó una alteración a los 90 minutos después de la dosis. No se observó una alteración de la memoria, según lo evaluado mediante la prueba de recordación inmediata y retardada de palabras a las 4 horas después de la dosis. Efecto rebote: En los estudios controlados de seguridad y eficacia, de 3 meses de duración (Estudio 1 y Estudio 2) se evaluó la presencia de insomnio de rebote luego de la discontinuación de Belsomra respecto del placebo y de la situación inicial en pacientes adultos no ancianos que recibieron 40 mg ó 20 mg de Belsomra y en pacientes de edad avanzada que recibieron 30 ó 15 mg de Belsomra. No se observaron efectos claros sobre las mediciones de aparición o mantención del sueńo. Efectos de deprivación: En los estudios controlados de seguridad y eficacia, de 3 meses de duración (Estudio 1 y Estudio 2) se evaluó la presencia de insomnio de rebote luego de la discontinuación de Belsomra respecto del placebo y de la situación inicial en pacientes adultos no ancianos que recibieron 40 mg o 20 mg de Belsomra y en pacientes de edad avanzada que recibieron 30 ó 15 mg de Belsomra. El análisis no mostró evidencia clara de deprivación en la población total del estudio sobre la base de la evaluación de las respuestas de los pacientes al Cuestionario de Tyrer de Síntomas de Deprivación o de la evaluación de eventos adversos relacionados con la deprivación luego de la discontinuación de Belsomra. Seguridad respiratoria: Uso en sujetos sanos con función respiratoria normal: Se evaluó el efecto depresor respiratorio de suvorexant (40 y 150 mg) después de 1 noche de tratamiento en un estudio cruzado, aleatorizado, controlado con placebo, doble ciego, en sujetos sanos no ancianos (n=12). A las dosis estudiadas, suvorexant no tuvo un efecto depresor respiratorio de acuerdo con la medición de la saturación de oxígeno (Véase Advertencias y Precauciones, Uso en poblaciones específicas). Farmacología clínica: Mecanismo de acción: Suvorexant es un antagonista muy selectivo, reversible, de alta afinidad, del receptor de la orexina en los receptores OX1R (Ki 0.55 nM) y OX2R (Ki 0.35 nM). El sistema de seńalización con el neuropéptido orexina es un promotor central de la vigilia. Los cuerpos celulares neuronales productores de orexina están ubicados específicamente en el hipotálamo y se proyectan hacia las neuronas del cerebro que median en el estado de vigilia. Suvorexant posibilita el proceso fisiológico por el cual las transiciones cerebrales de vigilia a sueńo mediante el bloqueo reversible de la unión de los neuropéptidos orexina A y orexina B, promotores de la vigilia, a OX1R y OX2#, suprimiendo así el impulso para despertar. Suvorexant no posee actividad farmacológica directa ni afinidad de unión (Ki >10 µM) a los receptores del ácido gamma aminobutírico (GABA), serotonina, dopamina, noradrenalina, melatonina, histamina, acetilcolina ni de opiáceos. Farmacodinamia: Evaluación del efecto de Belsomra sobre el intervalo QTc: Se evaluaron los efectos de suvorexant sobre el intervalo QTc en un estudio cruzado, aleatorizado, controlado con placebo y con agente activo (400 mg de moxifloxacino) en sujetos sanos (n=53). El límite superior del intervalo de confianza monolateral del 95% para el intervalo QTc corregido según el valor basal y ajustado según el placebo estuvo por debajo de los 10 ms, basado en el análisis de dosis de suvorexant de hasta 240 mg. Por tanto, suvorexant no prolonga el intervalo QTc en un grado que resulte clínicamente significativo. Farmacocinética: La exposición a suvorexant aumenta de una manera menos que estrictamente proporcional a la dosis dentro del rango de 10-80 mg debido a una disminución de la absorción. La farmacocinética de suvorexant es similar en sujetos sanos y en pacientes con insomnio. Absorción: Las concentraciones máximas de suvorexant se presentan a una tmax mediana de 2 horas (rango entre 30 minutos y 6 horas) bajo condiciones de ayuno. La biodisponibilidad absoluta promedio de 20 mg es del 62% (percentil 5to - 95to: 55% a 69%). La ingestión de suvorexant con una comida rica en grasa no resultó en un cambio significativo del AUC o Cmax, aunque se produjo un retardo de aproximadamente 1.5 horas en la tmax. Suvorexant puede tomarse con o sin alimento; sin embargo, para una aparición más rápida del sueńo, no se debiera administrar suvorexant con una comida o justo después de comer. Distribución: El valor promedio del volumen de distribución de suvorexant es de aproximadamente 49 litros. Suvorexant se une ampliamente (>99%) a las proteínas plasmáticas humanas, sin distribuirse preferentemente dentro de los eritrocitos. Suvorexant se une tanto la albúmina sérica humana como a la alfa-1 glicoproteína ácida. Metabolismo: Suvorexant se elimina principalmente a través del metabolismo, principalmente por medio de la CYP3A, con una contribución menor de parte de la CYP2C19. Las principales entidades circulantes son suvorexant y el metabolito hidroxi-suvorexant. No resulta esperable que este metabolito sea farmacológicamente activo. Eliminación: La ruta principal de eliminación es a través de las heces, con aproximadamente el 66% de la dosis radiomarcada recuperada en las heces comparado con el 23% recuperado en la orina. Suvorexant se elimina principalmente en forma de metabolitos, con menos del 1% de la dosis recuperada en las heces y orina como suvorexant. Los parámetros farmacocinéticos de suvorexant son lineales, con una acumulación de aproximadamente 1 a 2 veces con una administración 1 vez al día. El estado estacionario se alcanza a los 3 días luego de una administración 1 vez al día, lo que concuerda con una t1/2 promedio de aproximadamente 12 horas (CI del 95%: 12 a 13). Poblaciones especiales: Los efectos de la insuficiencia renal y hepática sobre la farmacocinética de suvorexant se evaluaron en estudios farmacocinéticos específicos. La exposición total a suvorexant (expresada como concentración total y no enlazada) resultó similar entre los pacientes con insuficiencia renal severa (clearance urinario de creatinina 30 ml/min/1.73 m2) y los sujetos control sanos equivalentes. No se requiere un ajuste de la dosis en pacientes con insuficiencia renal (ver Uso en poblaciones específicas). La exposición a suvorexant después de una dosis única fue similar en pacientes con insuficiencia hepática moderada (Child-Pugh categoría 7 a 9) y en sujetos control sanos equivalentes; sin embargo, la vida media terminal aparente de suvorexant aumentó de aproximadamente 15 horas (rango 10-22 horas) en sujetos sanos a aproximadamente 19 horas (rango 11-49 horas) en pacientes con insuficiencia hepática moderada. No se evaluó la farmacocinética de suvorexant en sujetos con insuficiencia hepática severa (Child-Pugh 10-15 puntos) (véase Advertencias y Precauciones, Uso en poblaciones específicas). La edad, género, IMC y raza fueron incluidos como factores evaluados en el modelo farmacocinético poblacional para evaluar la farmacocinética de suvorexant en sujetos sanos y para predecir las exposiciones en la población de pacientes. El género, IMC y raza no provocaron cambios clínicamente significativos en la farmacocinética de suvorexant. Un análisis de sujetos sanos no ancianos (< 65 ańos) y de edad avanzada (≥65 ańos) y de pacientes indica que la administración de dosis de 15 mg a los sujetos de edad avanzada proporciona exposiciones a suvorexant similares a las obtenidas con dosis de 20 mg en sujetos no ancianos (véase Posología, Uso en poblaciones específicas). En mujeres se observa un aumento del 17% y 9% en el AUC y Cmax, respectivamente, luego de la administración de 40 mg de Belsomra comparado con lo observado en hombres. La concentración promedio de suvorexant 9 horas después de la administración es 5% más alta en las mujeres en todo el rango de dosis estudiado (10-40 mg). No se justifica un ajuste de la dosis de Belsomra basado en el género. El clearance oral aparente de suvorexant está relacionado inversamente con el índice de masa corporal (IMC). En pacientes obesos, el AUC y la Cmax están aumentados en un 31% y 17%, respectivamente, comparado con los pacientes no obesos. La concentración promedio de suvorexant aproximadamente 9 horas después de una dosis de 20 mg es un 15% más alta en pacientes obesos (IMC > 30 kg/m2) respecto de aquellos con un IMC normal (IMC 25 kg/m2). No se justifica un ajuste de la dosis basado en el IMC. Interacciones medicamentosas: Fármacos activos a nivel del SNC: Se observó un efecto aditivo sobre el desempeńo psicomotor cuando una dosis única de 40 mg de suvorexant se administró concomitantemente con una dosis única de 0.70 g/kg de alcohol. Suvorexant no afectó las concentraciones de alcohol y el alcohol no afectó las concentraciones de suvorexant (ver Advertencias y Precauciones, Interacciones medicamentosas). Un estudio de interacciones con una dosis única de 40 mg de suvorexant y 20 mg de paroxetina a niveles de estado estacionario en sujetos sanos no demostró la existencia de una interacción farmacocinética o farmacodinámica clínicamente significativa (ver Advertencias y Precauciones, Interacciones medicamentosas. Efectos de otros medicamentos sobre suvorexant: Los efectos de otros medicamentos sobre la farmacocinética de suvorexant se presentan en la Figura 1 como el cambio respecto de suvorexant administrado como monoterapia (prueba/referencia). Los inhibidores potentes de la CYP3A (por ej., ketoconazol) aumentan de manera significativa la exposición a suvorexant. Los inhibidores moderados de la CYP3A (por ej., diltiazem) tienen efectos más bajos. Los inductores potentes de la CYP3A (por ej., rifampicina) reducen significativamente la exposición a suvorexant (ver Advertencias y precauciones, Interacciones medicamentosas).
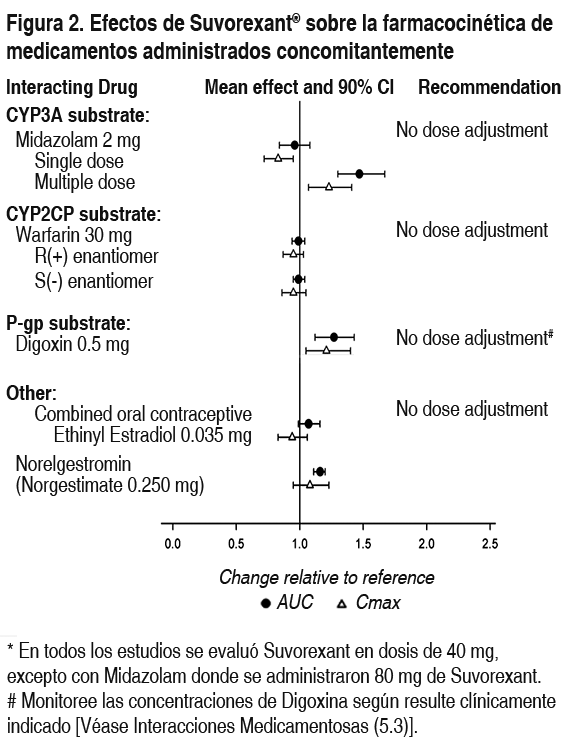
Efectos de suvorexant sobre otros medicamentos: Estudios del metabolismo in vitro demuestran que suvorexant posee el potencial de inhibir la CYP3A y la glicoproteína-P intestinal. Resulta improbable que suvorexant provoque una inhibición clínicamente significativa de las isoformas humanas CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19 o CYP2D6. Además, no se prevé una inhibición clínicamente significativa de los transportadores OATP1B1, BCRP y OCT2. Resulta improbable que la administración crónica de suvorexant induzca el metabolismo de medicamentos metabolizados por las principales isoformas CYP. En la Figura 2 se presentan los efectos específicos in vivo sobre la farmacocinética del midazolam, warfarina, digoxina y anticonceptivos orales como el cambio respecto del medicamento interactuante administrado como monoterapia (prueba/referencia) (véase Interacciones medicamentosas).
Toxicología animal: Toxicidad aguda: No se han realizado estudios de toxicidad con dosis única. En estudios con dosis múltiples no se observó mortalidad en ratas ni perros después de la primera dosis a exposiciones sistémicas de 88 veces y 59 veces, respectivamente, la exposición clínica a la Dosis Humana Máxima Recomendada (MRHD) (20 mg). Toxicidad crónica: En estudios a largo plazo en ratas, los hallazgos consistieron en cambios patológicos clínicos menores y alteraciones histomorfológicas en el hígado, tiroides, páncreas y estómago. Una hipertrofia hepatocelular y de las células foliculares en la tiroides fueron hallazgos concordantes con una inducción de las enzimas microsomales, la que está bien descrita como específica de los roedores. La inflamación crónica del páncreas en ratas macho resultó semejante al cambio pancreático relacionado con la edad observado en ratas viejas, y la erosión de la mucosa glandular en el estómago se consideró una toxicidad local del producto experimental o formulación. No se observaron efectos adversos en ratas a exposiciones de 19 a 23 veces la exposición clínica a la MRHD (20 mg). En el estudio de carcinogenicidad en ratas de 2 ańos de duración, la incidencia base de atrofia retinal inducida por la edad y la luz se apreció aumentada en los animales que recibieron dosis media y alta. Los AUC plasmáticos con la dosis sin efecto para la atrofia retinal fueron aproximadamente 7 veces (ratas macho) y 11 veces (ratas hembra) aquellos observados en seres humanos a la MRHD (20 mg). En un estudio posterior en rata se produjo un aumento paradójico en el tiempo necesario para despertar durante la fase con luz después de la administración de dosis suprafarmacológicas de un antagonista del receptor de la orexina. Además, en estudios de suvorexant en ratas albinas y pigmentadas, la atrofia retina mostró un retraso en su aparición y presentó una incidencia y severidad más bajas en ratas pigmentadas comparado con las ratas albinas, lo que indica que la pigmentación ocular protege al ojo contra el cambio a nivel ocular. No se observaron cambios retinales en los perros tratados con suvorexant durante 9 meses a exposiciones de hasta 84 veces aquella de los seres humanos a la MRHD (20 mg). Por lo tanto, el cambio retinal observado en el estudio de carcinogenicidad en rata parece corresponder a un cambio paradójico específico de las ratas y mediado farmacológicamente. En estudios a largo plazo en perros, los cambios patológicos clínicos adversos estuvieron circunscritos a aumentos relacionados con la dosis en la fosfatasa alcalina, y los hallazgos histomorfológicos estuvieron limitados a una hipertrofia hepatocelular que no se consideró adversa. No se observó toxicidad en perros a exposiciones de 45 veces la exposición clínica a la MRHD (20 mg).
Posología:Dosificación en adultos (<65 ańos) y adultos mayores (≥65 ańos): La dosis recomendada es de 10 mg, ingeridos no más de 1 vez por noche y dentro de los 30 minutos previos a acostarse, con al menos 7 horas más antes de la hora planificada para despertarse. Si la dosis de 10 mg es bien tolerada pero no efectiva, la dosis puede ser incrementada. La dosis máxima recomendada es de 20 mg 1 vez al día. Para los pacientes con enfermedad de Alzheimer de leve a moderada, el intervalo de dosis recomendado es de 10 a 20 mg tomados no más de 1 vez por noche y dentro de los 30 minutos de irse a la cama, con al menos 7 horas remanentes antes de la hora programada para despertarse. Uso con Inhibidores moderados de la CYP3A: Belsomra no está recomendado para pacientes que toman concomitante inhibidores fuertes o moderados de la CYP3A4 a medida que aumenta la exposición a suvorexant. Efecto del alimento: El tiempo hasta la aparición del efecto de Belsomra® podría verse retrasado si se toma con alimento o poco después haber comido. Poblaciones especiales: La exposición de Belsomra es mayor en obesos, cuando se compara con pacientes no obesos, y en mujeres comparadas con varones. Particularmente en obesos, el incremento del riesgo de eventos adversos relacionados con la exposición deberá ser considerado antes de incrementar la dosis. Uso con depresores del sistema nervioso central: Cuando Belsomra se usa combinado con otros depresores del sistema nervioso central, puede ser necesario ajustar la dosis de Belsomra y/o de los otros depresores, debido al potencial efecto aditivo.
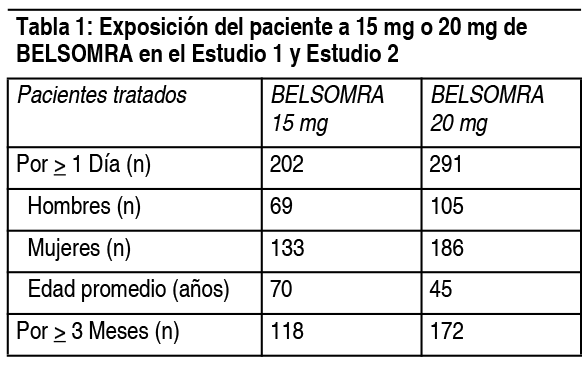
Efectos Colaterales:Estudios clínicos con experiencia en adultos: Debido a que los estudios clínicos se realizan bajo una amplia variedad de condiciones, las tasas de reacciones adversas observadas en los estudios de un fármaco no pueden compararse directamente con las tasas obtenidas en los estudios de otros fármacos y puede que no reflejen las tasas observadas en la práctica clínica. En estudios de eficacia controlados, de 3 meses de duración (Estudio 1 y Estudio 2), 1.263 pacientes fueron expuestos a Belsomra, incluyendo 493 pacientes que recibieron 15 mg o 20 mg de Belsomra (ver Tabla 1). En un estudio a largo plazo se trataron pacientes adicionales (n=521) con Belsomra a dosis mayores que las recomendadas, incluyendo un total de 160 pacientes que recibieron Belsomra por al menos 1 ańo.
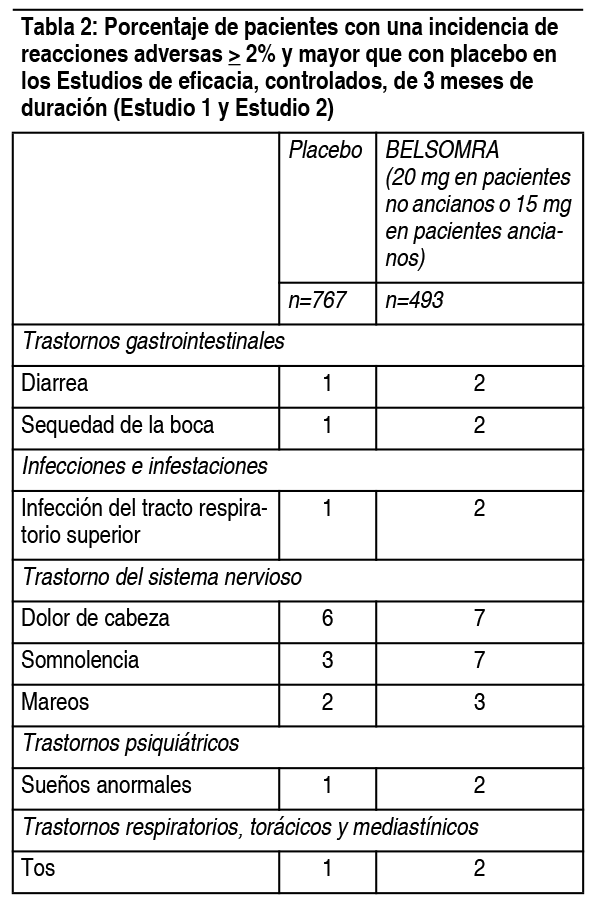
Los datos de seguridad combinados descritos a continuación (ver Tabla 2) reflejan el perfil de reacciones adversas durante los primeros 3 meses de tratamiento. Reacciones adversas que resultan en la discontinuación del tratamiento: La incidencia de discontinuación debido a reacciones adversas para pacientes tratados con 15 mg o 20 mg de Belsomra fue de 3% comparado con un 5% para placebo. Ninguna reacción adversa individual condujo a la discontinuación con una incidencia ≥1%. Reacciones adversas observadas en estudios clínicos controlados con placebo: La Tabla 2 muestra el porcentaje de pacientes con reacciones adversas durante los primeros 3 meses de tratamiento, basado en los datos combinados de los estudios de eficacia controlados, de 3 meses de duración (Estudio 1 y Estudio 2). El perfil de reacciones adversas en pacientes de edad avanzada fue generalmente concordante con el de los pacientes no ancianos. Las reacciones adversas informadas durante un tratamiento a largo plazo de hasta 1 ańo fueron generalmente concordantes con aquellas observadas durante los primeros 3 meses de tratamiento.
En los estudios clínicos se ha reportado parálisis del sueńo y alucinaciones hipnagógicas/hipnopómpicas en pacientes que toman Belsomra (<1%). Exámenes de laboratorio: En estudios controlados con placebo en pacientes con insomnio, no se observaron diferencias clínicamente significativas en los parámetros habituales de química sérica, hematología o análisis de orina, entre los pacientes que recibieron Belsomra y aquellos que recibieron placebo. Pacientes con enfermedad de Alzheimer leve a moderada: En un estudio de 4 semanas en pacientes con enfermedad de Alzheimer leve a moderada que recibieron Belsomra, el perfil de reacciones adversas fue generalmente similar al perfil de los estudios pivotales de seguridad (ver Tabla 2). Experiencia post-comercialización: Las siguientes reacciones adversas han sido identificadas durante el uso posterior a la aprobación de Belsomra. Debido a que estas reacciones son reportadas voluntariamente por una población de tamańo incierto, no siempre es posible estimar de manera fiable su frecuencia o establecer una relación causal con la exposición al fármaco. Trastornos cardíacos: palpitaciones, taquicardia. Transtornos gastrointestinales: Náuseas, vómito. Trastornos generales y condiciones del sitio de administración: sensación anormal. Trastornos del sistema nervioso: hiperactividad psicomotora. Trastornos psiquiátricos: pesadillas. Trastornos de la piel y del tejido subcutáneo: prurito.
Contraindicaciones: Belsomra está contraindicado en pacientes con una conocida hipersensibilidad a suvorexant o a cualquiera de sus excipientes. Belsomra no debe ser usado en pacientes que sufren narcolepsia.
Advertencias:Pacientes con narcolepsia o cataplejía: No se ha estudiado el uso de Belsomra en pacientes con narcolepsia o cataplejía. Por lo tanto, no se recomienda el tratamiento con Belsomra en estos pacientes. Efectos depresores del SNC: Belsomra es un depresor del sistema nervioso central (SNC) que puede alterar la vigilia durante las horas de luz de día, incluso cuando se usa según lo indicado. Los profesionales que lo indican debieran efectuar un seguimiento en busca de somnolencia y efectos depresores del SNC. Belsomra puede alterar las habilidades para conducir y aumentar el riesgo de quedarse dormido mientras se conduce. Los pacientes deben evitar involucrarse en actividades peligrosas, como usar un vehículo motorizado o maquinaria pesada, mientras se encuentren tomando Belsomra. Se debiera precaver a los pacientes respecto de la posible alteración del desempeńo de dichas actividades que puede ocurrir al día siguiente de la ingestión (véase Estudios clínicos). Se debiera aconsejar a los pacientes que no consuman alcohol en combinación con Belsomra debido a los efectos aditivos (véase Interacciones medicamentosas). El riesgo de alteración al día siguiente, incluyendo problemas para conducir, se ve aumentado si Belsomra se toma cuando resta menos de una noche completa de sueńo, si se toma una dosis mayor que la recomendada, si se administra concomitantemente con otros depresores del SNC, o si se administra conjuntamente con otros medicamentos que aumentan los niveles sanguíneos de Belsomra. No se recomienda el uso de Belsomra con otros medicamentos para tratar el insomnio. Necesidad de evaluar diagnósticos comórbidos: Puesto que los trastornos del sueńo pueden ser la manifestación presente de un trastorno físico y/o psiquiátrico, el tratamiento sintomático del insomnio debiera iniciarse sólo después de una cuidadosa evaluación del paciente. El agravamiento del insomnio o la aparición de nuevas alteraciones cognitivas o conductuales podrían ser el resultado de una alteración psiquiátrica o física subyacente no reconocida, diferente del insomnio, la que debiera ser evaluada. Pensamiento anormal y cambios conductuales: Se ha informado la aparición de una diversidad de cambios cognitivos y conductuales con el uso de hipnóticos como el Belsomra. Se han reportado conductas complejas tales como "conducir dormido" (es decir, conducir mientras no se está completamente despierto después de la ingestión de un hipnótico) y otras conductas complejas (por ejemplo, caminar dormido, o preparar y comer alimento mientras se está dormido), con amnesia para el evento, en asociación con el uso de hipnóticos. Estos eventos se pueden presentar en personas que nunca antes usaron hipnóticos como también en aquellos que los han usado anteriormente. El uso de alcohol y otros depresores del SNC pueden aumentar el riesgo de estas conductas. Se debe considerar encarecidamente la discontinuación de Belsomra en aquellos pacientes que informan cualquier conducta compleja relacionada con el sueńo (véase Interacciones farmacológicas). Agravamiento de la depresión/ideación suicida: En pacientes con depresión primaria se ha informado un agravamiento de la depresión, incluso pensamientos y actos suicidas (incluyendo suicidios realizados), en asociación con el uso de hipnóticos. La aparición de cualquier signo o síntoma conductual nuevo preocupante requiere de una evaluación cuidadosa e inmediata. Pacientes con compromiso de la función respiratoria: No se ha estudiado el uso de Belsomra en pacientes con apnea obstructiva del sueńo (AOS) severa o enfermedad pulmonar obstructiva crónica (EPOC). Se aconseja precaución al indicar Belsomra a pacientes con compromiso de la función respiratoria (ver Uso en poblaciones específicas). Inhibidores potentes de la CYP3A: No se recomienda el uso concomitante de Belsomra con inhibidores potentes de la CYP3A (véase Interacciones medicamentosas). Pacientes con insuficiencia hepática severa: No se ha estudiado el uso de Belsomra en pacientes con insuficiencia hepática severa y no se recomienda para estos pacientes (ver Uso en poblaciones específicas).
Precauciones:Uso en poblaciones específicas: Embarazo: No se han realizado estudios clínicos en embarazadas. Se debiera utilizar Belsomra durante el embarazo sólo si el beneficio potencial justifica el posible riesgo para el feto. Madres nodrizas: No se sabe si suvorexant se secreta en la leche materna; no obstante, suvorexant y el metabolito hidroxi-suvorexant se secretaron en la leche de ratas nodrizas en niveles mayores (9 y 1.5 veces, respectivamente) que aquellos observados en el plasma materno. Debido a que muchos fármacos se excretan en la leche materna humana, se debe tener cuidado al momento de administrar a mujeres nodrizas. Uso pediátrico: No se han determinado la seguridad y eficacia de Belsomra en pacientes pediátricos. Por lo tanto, no se recomienda el tratamiento con Belsomra. Uso geriátrico: Del número total de pacientes tratados con Belsomra (N=1.784) en estudios clínicos de fase 3 para investigar la seguridad y eficacia, 829 pacientes tenían 65 ańos o más, mientras que 159 tenían 75 ańos o más. No se observaron diferencias clínicamente significativas en la seguridad o eficacia entre estos pacientes y aquellos más jóvenes a las dosis recomendadas. No obstante, no se puede descartar una sensibilidad mayor en algunos sujetos de edad avanzada (véase Posología, Estudios clínicos, Farmacología clínica). Pacientes con enfermedad de Alzheimer leve a moderada: En un estudio de 4 semanas controlado con placebo en pacientes (50 ańos y más) con enfermedad de alzheimer de leve a moderada, no se observaron diferencias clínicamente significativas en la seguridad en pacientes tratados con Belsomra en comparación con pacientes tratados con placebo, incluidas las evaluaciones de comportamiento cognitivo y neuropsiquiátrico (ver Efectos colaterales; Estudios clínicos). Pacientes con compromiso de la función respiratoria: Se debieran tomar en consideración los efectos de Belsomra sobre la función respiratoria si se indica a pacientes con compromiso de la función respiratoria. Apnea obstructiva del sueńo: Se evaluó el efecto depresor respiratorio de Belsomra después de 1 noche y luego de 4 noches seguidas de tratamiento en un estudio cruzado, aleatorizado, controlado con placebo, de 2 períodos, en pacientes (n=26) con apnea obstructiva del sueńo (AOS) leve a moderada. Luego de dosis de 40 mg 1 vez al día, el promedio del índice de apnea/hipopnea con la diferencia de tratamiento (suvorexant - placebo) el día 4 fue de 2.7 (CI del 90%: 0.22 a 5.09). No se pueden excluir los efectos respiratorios clínicamente significativos de Belsomra en pacientes con AOS. No se ha estudiado el uso de Belsomra en pacientes con AOS severa (véase Advertencias y precauciones). Enfermedad pulmonar obstructiva crónica: Se evaluó el efecto depresor respiratorio de Belsomra después de 1 noche y luego de 4 noches seguidas de tratamiento en un estudio cruzado, aleatorizado, controlado con placebo, de 2 períodos, en pacientes (n=25) con enfermedad pulmonar obstructiva crónica (EPOC) leve a moderada. Belsomra (40 mg para sujetos no ancianos, 30 mg en sujetos de edad avanzada) no tuvo efectos depresores respiratorios en pacientes con EPOC leve a moderada, de acuerdo con la medición de la saturación de oxígeno. No se pueden excluir los efectos respiratorios clínicamente significativos de Belsomra en pacientes con EPOC. No se ha estudiado el uso de Belsomra en pacientes con EPOC severa (véase Advertencias y Precauciones). Pacientes con insuficiencia hepática: La farmacocinética de suvorexant fue similar en pacientes con insuficiencia hepática moderada y en sujetos sanos. No se requiere un ajuste de la dosis en pacientes con insuficiencia hepática leve y moderada. No se ha estudiado el uso de Belsomra en pacientes con insuficiencia hepática severa y no se recomienda para estos pacientes (véase Advertencias y Precauciones, Farmacología clínica). Pacientes con insuficiencia renal: La farmacocinética de suvorexant fue similar en pacientes con insuficiencia renal severa y en sujetos sanos. No se requiere un ajuste de la dosis en pacientes con insuficiencia renal (véase Farmacología clínica). Carcinogénesis: No se observó evidencia de potencial carcinogénico en ratones hemicigotos Tg.rasH2 que recibieron dosis por vía oral durante 6 meses a exposiciones de hasta 105 veces la exposición clínica a la MRHD (20 mg). En el estudio de carcinogenicidad en ratas de 3 ańos de duración se observó un aumento de la incidencia de adenomas en las células foliculares hepáticas y tiroídeas. Estos cambios fueron secundarios a una inducción de las enzimas hepáticas y un aumento de la producción de TSH, respectivamente, mecanismos que se encuentran bien descritos como específicos de roedores. A la dosis sin efecto para tumores, la exposición fue 7 veces la exposición clínica a la MRHD (20 mg). Mutagénesis: Suvorexant resultó negativo en los ensayos toxicológicos genéticos in vitro (mutación bacteriana inversa, elución alcalina y aberración cromosómica) e in vivo (micronúcleo de rata y ratón). Reproducción: En ratas macho y hembra tratadas antes y durante el apareo y gestación temprana se observaron disminuciones en el cuerpo lúteo, implantaciones y fetos vivos resultantes. No hubo efectos en los machos. A la dosis sin efecto, la exposición fue aproximadamente 20 veces la exposición clínica a la MRHD (20 mg). Desarrollo: No se observó teratogenicidad en ratas o conejas preńadas tratadas durante el período de organogénesis. En ratas, la toxicidad durante el desarrollo se limitó a una disminución en los pesos corporales fetales. En conejos no se observó evidencia de toxicidad durante el desarrollo. La exposición materna en ratas y conejos a la dosis sin efecto fue de 25 veces y 39 veces la exposición clínica a la MRHD (20 mg), respectivamente. Luego de la administración oral a ratas durante toda la gestación y lactancia, los efectos sobre la prole estuvieron circunscritos a una disminución transitoria del peso corporal con la dosis más alta examinada. La exposición materna a la dosis sin efecto fue 25 veces la exposición clínica a la MRHD (20 mg).
Interacciones Medicamentosas:Interacciones medicamentosas y otras formas de interacción: Agentes activos a nivel del SNC: Cuando se administró Belsomra concomitantemente con alcohol se demostró una alteración psicomotora aditiva. No se observó una alteración en la farmacocinética de ninguna de las 2 drogas (véase Advertencias y Precauciones, Farmacología clínica). Cuando se administró Belsomra concomitantemente con paroxetina no se observó una interacción farmacocinética o farmacodinámica clínicamente significativa (véase Advertencias y Precauciones, Farmacología clínica). Efectos de otros medicamentos sobre suvorexant: El metabolismo por parte de la CYP3A es la principal ruta de eliminación para suvorexant. Inhibidores de la CYP3A: La administración concomitante de suvorexant con ketoconazol (un potente inhibidor de la CYP3A) aumentó de manera importante la exposición a suvorexant. No se recomienda el uso concomitante de Belsomra con inhibidores potentes de la CYP3A (por ej., ketoconazol, itraconazol, posaconazol, claritromicina, nefazodona, ritonavir, saquinavir, nelfinavir, indinavir, boceprevir, telaprevir, telitromicina y conivaptan) (véase Advertencias y Precauciones, Farmacología clínica). La administración concomitante de suvorexant con diltiazem (un potente moderado de la CYP3A) aumentó la exposición a suvorexant. La dosis recomendada de Belsomra es 10 mg, la que no debiera sobrepasarse cuando se administra conjuntamente con inhibidores moderados de la CYP3A (por ej., amprenavir, aprepitant, atazanavir, ciprofloxacino, diltiazem, eritromicina, fluconazol, fosamprenavir, jugo de pomelo, crizotinib, imatinib y verapamilo) (véase Posología, Advertencias y Precauciones y Farmacología clínica). Inductores de la CYP3A: La exposición a suvorexant puede verse disminuida en un grado importante cuando se administra concomitantemente con inductores potentes de la CYP3A (por ej., rifampicina, carbamazepina y fenitoína). Es posible que se produzca una reducción en la eficacia de suvorexant (véase Farmacología clínica). Efectos de suvorexant sobre otros medicamentos: Suvorexant es un débil inhibidor de la CYP3A y del transportador glicoproteína-P intestinal luego de la administración seguida de dosis múltiples. Midazolam: La administración concomitante de suvorexant con midazolam (un substrato sensible a la CYP3A) aumentó levemente la exposición al midazolam. En el caso de la mayoría de los fármacos metabolizados por la CYP3A no resulta esperable que suvorexant aumente las concentraciones plasmáticas en un grado clínicamente significativo (véase Farmacología clínica). Digoxina: La administración concomitante de suvorexant con digoxina aumentó levemente los niveles de digoxina debido a la inhibición de la glicoproteína-P intestinal. Se considera clínicamente indicado el seguimiento de las concentraciones de digoxina cuando se administra concomitantemente con Belsomra (véase Farmacología clínica). Otra terapia concomitante: No se observaron interacciones farmacocinéticas clínicamente significativas luego de la administración concomitante de suvorexant con warfarina o anticonceptivos orales (véase Farmacología clínica).
Sobredosificación:Abuso y dependencia a drogas: Abuso: En un estudio de propensión al abuso (consumo excesivo) conducido en usuarios de múltiples drogas recreativas (N=36), suvorexant (40, 80 y 150 mg) produjo efectos similares a aquellos del zolpidem (15, 30 mg) en las clasificaciones subjetivas de "sensación placentera con la droga". Al igual que con otros hipnóticos, se debe tener precaución al momento de recetar Belsomra a sujetos con antecedentes de adición, o consumo excesivo (abuso), de drogas o alcohol debido al riesgo de un uso incorrecto o abuso. Dependencia: En estudios clínicos finalizados con Belsomra no se observó evidencia de dependencia física con el uso prolongado de Belsomra. No se encontró evidencia de síntomas de deprivación después de la discontinuación de Belsomra. Sobredosificación: Existe una limitada experiencia clínica previa a la comercialización con los efectos de una sobredosis de Belsomra. En estudios de farmacología clínica se administró a sujetos sanos dosis de hasta 240 mg de suvorexant en la mańana, observándose un aumento, dependiente de la dosis, en la frecuencia y duración de la somnolencia reportada. Se debieran usar medidas generales sintomáticas y de apoyo, junto con un lavado gástrico inmediato cuando corresponda. En caso de necesidad se debieran administrar líquidos por vía intravenosa. Como en todos los casos de sobredosis de droga, se debiera realizar un seguimiento de la respiración, pulso, presión arterial y otros signos vitales adecuados, y emplear medidas de apoyo generales. No se ha determinado el valor de la diálisis en el tratamiento de la sobredosis. Como suvorexant se enlaza en gran medida a las proteínas, no resulta esperable que la hemodiálisis contribuya a la eliminación de suvorexant. Como ocurre con el manejo de todas las sobredosis, se debiera considerar la posibilidad de la ingestión de múltiples drogas. Es posible que el profesional sanitario desee considerar la posibilidad de contactar a un centro de intoxicaciones para acceder a información actualizada acerca del manejo de una sobredosis con un medicamento hipnótico.
Conservación:Almacenamiento: Almacenar a no más de 25° C. Precauciones especiales de almacenamiento: Mantener en su envase original hasta su uso, protegido de la humedad y de la luz.
Presentaciones: Estuche de cartulina impreso que contiene 30 comprimidos recubiertos.
 Laboratorio
Laboratorio