Composición: Cada comprimido recubierto contiene: 100 mg de Doravirina, 300 mg de Lamivudina y 300 mg de Tenofovir Disoproxilo Fumarato equivalente a 245 mg de Tenofovir Disoproxilo. Excipiente con efecto conocido: Cada comprimido recubierto contiene 8.6 mg de Lactosa (como monohidrato).Forma farmacéutica: Comprimido recubierto. Lista de excipientes: Núcleo del comprimido: Croscarmelosa Sódica; Hipromelosa Succinato; Acetato; Estearato de Magnesio; Celulosa Microcristalina; Dióxido de Silicio Coloidal; Estearil Fumarato de Sodio. Recubrimiento: Cera de Carnauba, Hipromelosa; Óxido de Hierro Amarillo; Lactosa Monohidrato; Dióxido de Titanio Triacetina.
Acción Terapéutica:Grupo farmacoterapéutico: Combinaciones de antirretroviarles para el tratamiento de la infección por VIH. Código ATC: J05AR24.
Indicaciones: Delstrigo está indicado para el tratamiento de adultos infectados por el VIH-1 sin evidencia pasada o presente de resistencia a inhibidores de la transcriptasa inversa no nucleósidos (ITINN), lamivudina o tenofovir (ver las secciones Advertencias y precauciones especiales de empleo y Propiedades farmacodinámicas).
Propiedades:Propiedades farmacodinámicas: Mecanismo de acción: Doravirina: Doravirina es un inhibidor de la transcriptasa inversa del VIH-1 no nucleósido derivado de la piridinona e inhibe la replicación del VIH-1 mediante la inhibición no competitiva de la transcriptasa inversa (TI) del VIH-1. Doravirina no inhibe las ADN polimerasas celulares α, ß ni la ADN polimerasa γ mitocondrial del ser humano. Lamivudina: Lamivudina es un análogo nucleósido. En el interior de la célula, lamivudina se fosforila a su metabolito activo 5´-trifosfato, lamivudina trifosfato (3TC-TP). El mecanismo de acción principal de 3TC-TP es la inhibición de la TI mediante la terminación de la cadena de ADN tras la incorporación del análogo nucleotídico. Tenofovir disoproxilo: Tenofovir disoproxilo es un diéster fosfonato de un análogo nucleósido acíclíco de la adenosina monofosfato. Tenofovir disoproxilo requiere una hidrólisis inicial del diéster para su conversión a tenofovir y posteriores fosforilaciones por acción de enzimas celulares para formar difosfato de tenofovir. El difosfato de tenofovir inhibe la actividad de la TI del VIH-1 al competir con el sustrato natural desoxiadenosina 5´-trifosfato tras incorporarse al ADN formando parte de la terminación de la cadena. El difosfato de tenofovir es un inhibidor débil de las ADN polimerasas α y β y la ADN polimerasa γ mitocondrial de mamíferos. Actividad antiviral en cultivo celular: Doravirina: Doravirina mostró un valor de CE50 de 12.0 ± 4.4 nM frente a cepas de laboratorio naturales del VIH 1 cuando se analizó en presencia de suero humano normal al 100% con células indicadoras de MT4-GFP. Doravirina mostró actividad antiviral frente a un amplio grupo de cepas primarias de VIH- 1 (A, A1, AE, AG, B, BF, C, D, G, H) con valores de CE50 comprendidos entre 1.2 nM y 10.0 nM. La actividad antiviral de doravirina no fue antagonista cuando se combinó con lamivudina y tenofovir disoproxilo. Lamivudina: La actividad antiviral de lamivudina contra el VIH-1 se evaluó en una serie de estirpes celulares, como monocitos y células mononucleares de sangre periférica (CMSP), utilizando análisis de sensibilidad convencionales. Los valores de EC50 estuvieron en el intervalo de 0.003 a 15 microM (1 microM = 023 mcg por ml). La mediana de los valores CE50 de lamivudina fueron de 60 nM (intervalo: 20 a 70 nM), 35 nM (intervalo: 30 a 40 nM), 30 nM (intervalo: 20 a 90 nM), 20 nM (intervalo: 3 a 40 nM), 30 nM (intervalo: 1 a 60 nM), 30 nM (intervalo: 20 a 70 nM), 30 nM (intervalo: 3 a 70 nM) y 30 nM (intervalo: 20 a 90 nM) frente a los virus de los subtipos A-G y el grupo O del VIH-1 (n=3 excepto n=2 para el subtipo B), respectivamente. La ribavirina (50 microM) utilizada en el tratamiento de la infección crónica por el VHC redujo la actividad anti-VIH-1 de lamivudina en 3.5 veces en las células MT-4. Tenofovir disoproxilo: La actividad antiviral de tenofovir frente a cepas del VIH-1 aisladas en el laboratorio y en la práctica clínica se evaluó en estirpes celulares de linfoblastos T, monocitos/macrófagos primarios y linfocitos de sangre periférica. Los valores CE50 de tenofovir estuvieron en el intervalo de 0.04-8.5 microM. Tenofovir mostró actividad antiviral en cultivo celular frente a los subtipos A, B, C, D, E, F, G y O del VIH-1 (los valores de CE50 variaron entre 0.5 y 2.2 microM). Resistencia en cultivo celular: Doravirina: Se seleccionaron cepas resistentes a doravirina en cultivo celular a partir de VIH-1 natural de diferentes orígenes y subtipos, así como de VIH-1 resistente a ITINN. Las sustituciones de aminoácidos emergentes observadas en la TI fueron: V106A, V106M, V106I, V108I, F227L, F227C, F227V, H221Y, M230I, L234I, P236L e Y318F. En el estudio in vitro no se seleccionaron mutaciones con resistencia a ITINN frecuentes (K103N, Y181C). La sustitución V106A (con la que se apreció una variación cercana a 19 veces) apareció como sustitución inicial en virus del subtipo B y las sustituciones V106A o M en virus de los subtipos A y C. Posteriormente, aparecieron sustituciones de F227(L/C/V) o L234I además de la sustitución V106 (mutaciones dobles que depararon una variación > 100 veces). Lamivudina: Se han seleccionado variantes del VIH-1 resistentes a lamivudina en cultivo celular y en pacientes tratados con lamivudina. El análisis genotípico mostró que la resistencia se debía a una sustitución específica de aminoácidos en la TI del VIH-1 en el codón 184, con un cambio de metionina por isoleucina o valina (M184V/I). Tenofovir disoproxilo: Las cepas del VIH-1 seleccionadas por tenofovir expresaron una sustitución K65R en la TI del VIH-1 y mostraron una disminución de la sensibilidad a tenofovir de 2-4 veces. Además, tenofovir seleccionó una sustitución K70E en la TI del VIH-1, lo que se asocia a una disminución de la sensibilidad a abacavir, emtricitabina, lamivudina y tenofovir a un nivel bajo. En ensayos clínicos: Pacientes adultos no tratados previamente: Doravirina: Los ensayos de fase 3 DRIVE-FORWARD y DRIVE-AHEAD, que incluían pacientes no tratados previamente (n=747) donde las siguientes sustituciones de ITINN fueron parte de los criterios de exclusión: L100I, K101E, K101P, K103N, K103S, V106A, V106I, V106M, V108I, E138A, E138G, E138K, E138Q, E138R, V179L, Y181C,Y181I, Y181V, Y188C, Y188H, Y188L, G190A, G190S, H221Y, L234I, M230I, M230L, P225H, F227C, F227L, F227V. Se observó la siguiente resistencia de novo en el subgrupo de análisis de la resistencia (pacientes con una carga de ARN del VIH-1 superior a 400 copias por ml en el momento del fracaso virológico o en el momento de la retirada temprana del estudio y que disponían de datos de resistencia).
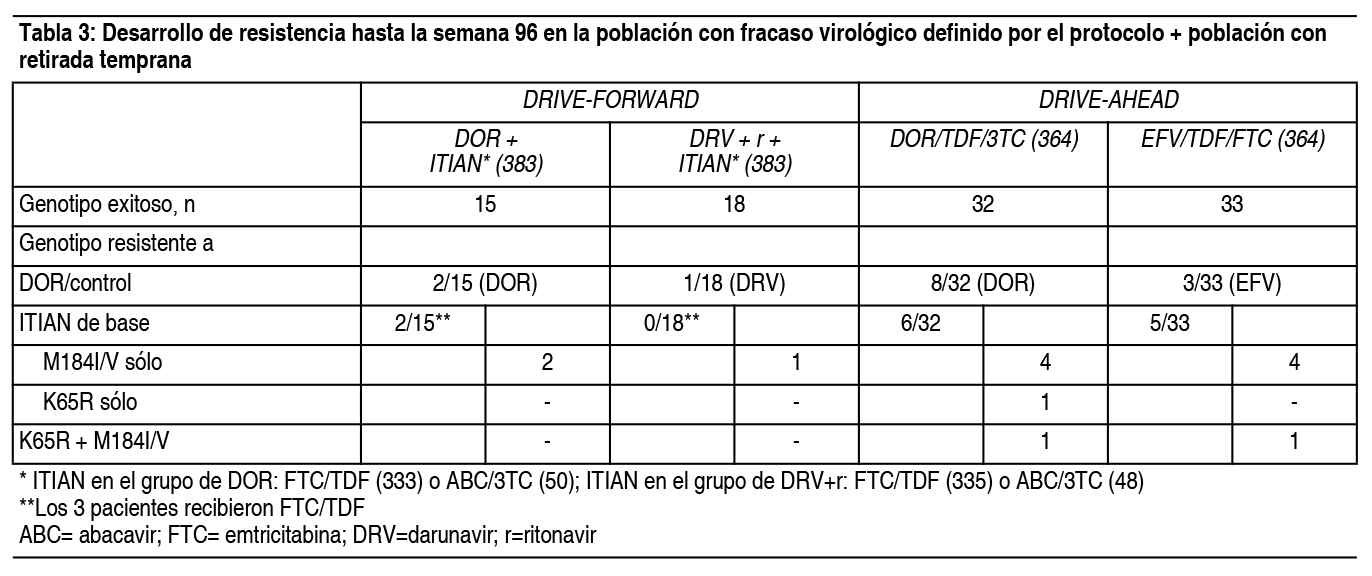
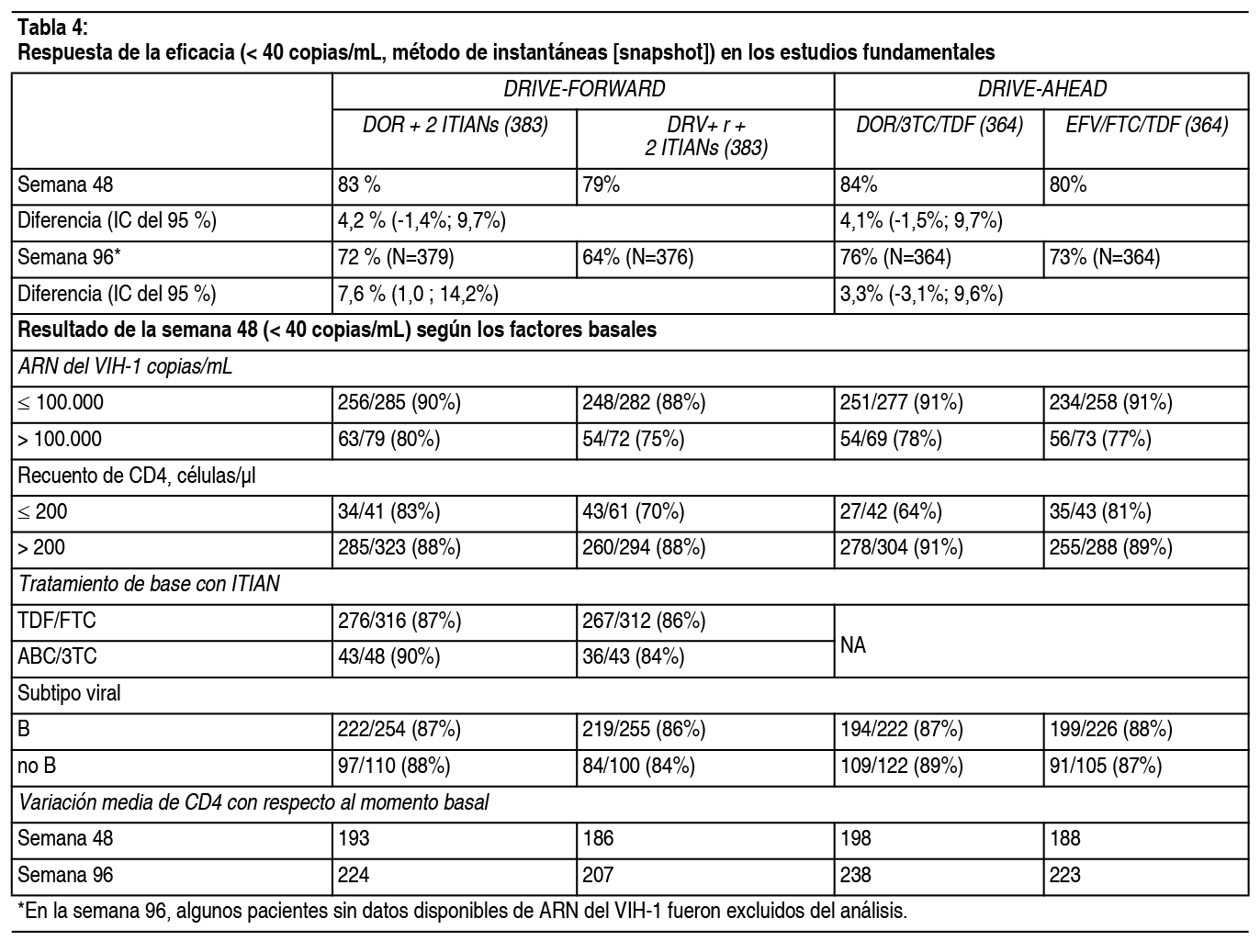
Las sustituciones aparecidas en la TI con resistencia asociada a doravirina fueron una o más de las siguientes: A98G, V106I, V106A, V106M/T, Y188L, H221Y, P225H, F227C, F227C/R y Y318Y/F. Pacientes adultos virológicamente suprimidos: El ensayo clínico DRIVE-SHIFT incluyó pacientes virológicamente suprimidos (N=670) sin antecedentes de fracaso al tratamiento (ver la sección, Experiencia clínica). Una ausencia documentada de resistencia genotípica a doravirina, lamivudina y tenofovir (antes de iniciar el primer tratamiento) fue parte de los criterios de inclusión para pacientes que cambiaron su tratamiento desde pautas posológicas basadas en un IP o en un inhibidor de la integrasa (INI). Las sustituciones ITINN excluyentes fueron las enumeradas anteriormente (DRIVE-FORWARD y DRIVE-AHEAD) con la excepción de las sustituciones RT K103N, G190A e Y181C (aceptadas en DRIVE-SHIFT). No se requirió la documentación del genotipado de la resistencia antes del tratamiento para aquellos pacientes que cambiaron el tratamiento desde una pauta posológica basada en ITINN. En el ensayo clínico DRIVE-SHIFT, ningún paciente desarrolló resistencia genotípica o fenotípica a DOR, 3TC o TDF en las primeras 48 semanas (cambio de tratamiento inmediato, N=447) o en las primeras 24 semanas (cambio de tratamiento retrasado, N=209) de tratamiento con Delstrigo. Un paciente desarrolló la mutación RT M184M/I y resistencia fenotípica a 3TC y FTC durante el tratamiento con su pauta posológica inicial. Ninguno de los 24 pacientes (11 del grupo de cambio de tratamiento inmediato, 13 del grupo de cambio de tratamiento retrasado) con mutaciones de ITINN iniciales (RT K103N, G190A o Y181C) experimentó fallo virológico hasta la semana 48 o en el momento de la interrupción del tratamiento. Resistencia cruzada: No se ha demostrado resistencia cruzada significativa entre las variantes del VIH-1 resistentes a doravirina y las resistentes a lamivudina/emtricitabina o tenofovir o entre las variantes resistentes a lamivudina o las resistentes a tenofovir y doravirina. Doravirina: Doravirina se evaluó en un número limitado de pacientes con resistencia a ITINN (K103N n=7, G190A n=1); todos los pacientes fueron suprimidos a <40 copias/ml en la semana 48. No se ha establecido un punto de corte para una reducción en la sensibilidad, producido por varias sustituciones de ITINN, que se asocie con una reducción en la eficacia clínica. Las cepas de laboratorio del VIH-1 que albergan las mutaciones frecuentes K103N e Y181C asociadas a ITINN o sustituciones de K103N/Y181C en la TI exhiben una disminución inferior a 3 veces de la sensibilidad a doravirina en comparación con el virus natural cuando se evalúan en presencia de suero humano normal al 100%. En los estudios in vitro, doravirina fue capaz de suprimir las siguientes sustituciones asociadas a ITINN: K103N, Y181C y G190A en concentraciones clínicamente relevantes. Se evaluó la sensibilidad a doravirina en presencia de suero bovino fetal al 10% en un grupo de 96 cepas clínicas diversas que contenían mutaciones asociadas a ITINN. Las cepas clínicas que contenían la sustitución Y188L o sustituciones de V106 en combinación con A98G, H221Y, P225H, F227C o Y318F mostraron una disminución de la sensibilidad a doravirina superior a100 veces. Otras sustituciones depararon una variación de 5-10 veces (G190S [5.7], K103N/P225H [7.9], V108I/Y181C [6.9], Y181V [5.1]). Se desconoce la importancia clínica de la disminución de la sensibilidad de 5-10 veces. Las sustituciones asociadas a resistencia a doravirina aparecidas durante el tratamiento pueden conferir resistencia cruzada a efavirenz, rilpivirina, nevirapina y etravirina. De los 7 pacientes que presentaron un elevado grado de resistencia a doravirina en los estudios fundamentales, 6 presentaron resistencia fenotípica a efavirenz y nevirapina, 3 a rilpivirina y 2 resistencia parcial a etravirina según el análisis Phenosense de Monogram. Lamivudina: Se ha observado resistencia cruzada entre ITIAN. La sustitución M184I/V de resistencia a lamivudina confiere resistencia a emtricitabina. Los mutantes del VIH-1 resistentes a lamivudina presentaron también resistencia cruzada a la didanosina (ddI). En algunos pacientes tratados con zidovudina más didanosina, han aparecido cepas resistentes a varios inhibidores de la TI, entre ellos lamivudina. Tenofovir disoproxilo: Se ha observado resistencia cruzada entre ITIAN. La sustitución K65R en la TI del VIH-1 seleccionada por tenofovir es también seleccionada en algunos pacientes infectados por el VIH-1 que reciben tratamiento con abacavir o didanosina. Cepas del VIH-1 con la sustitución K65R mostraron asimismo sensibilidad reducida a emtricitabina y lamivudina. Por consiguiente, se puede producir resistencia cruzada entre estos ITIAN en pacientes cuyos virus albergan la sustitución K65R. La sustitución K70E seleccionada clínicamente por tenofovir disoproxilo reduce la sensibilidad a abacavir, didanosina, emtricitabina, lamivudina y tenofovir. Cepas del VIH-1 aisladas en pacientes (N=20) cuyo VIH-1 expresaba una media de 3 sustituciones de aminoácidos en la TI asociadas a zidovudina (M41L, D67N, K70R, L210W, T215Y/F o K219Q/E/N) mostraron una disminución de la sensibilidad a tenofovir de 3.1 veces. Los pacientes cuyos virus expresaban una sustitución L74V de la TI, sin sustituciones asociadas a resistencia a zidovudina (N=8), tuvieron una respuesta reducida a tenofovir disoproxilo. Se dispone de datos limitados de pacientes cuyos virus expresaban una sustitución Y115F (N=3), una sustitución Q151M (N=2) o una inserción T69 (N=4) en la TI del VIH-1, todos ellos tuvieron una respuesta reducida en los ensayos clínicos. Experiencia clínica: Pacientes adultos no tratados previamente: La eficacia de doravirina se basa en los análisis de los datos de 96 semanas de dos ensayos de fase 3 aleatorizados, multicéntricos, doble ciego y controlados con tratamiento activo (DRIVE-FORWARD y DRIVE-AHEAD) en pacientes infectados por el VIH-1 no tratados previamente con un antirretroviral (n=1.494). Consultar en la sección Resistencia, las sustituciones de ITINN que formaban parte de los criterios de exclusión. En el ensayo DRIVE-FORWARD, 766 pacientes fueron aleatorizados y recibieron al menos 1 dosis de 100 mg de doravirina o 800 + 100 mg de darunavir + ritonavir 1 vez al día, cada uno de ellos en combinación con emtricitabina/tenofovir disoproxilo (FTC/TDF) o abacavir/lamivudina (ABC/3TC), a elección del investigador. En el momento basal, la mediana de edad de los pacientes era de 33 ańos (intervalo de 18 a 69 ańos); el 86% tenía un recuento de linfocitos T CD4+ mayor de 200 células/mm3, el 84% eran varones, el 27% eran de raza no blanca, el 4 % tenían coinfección por los virus de la hepatitis B o C, el 10 % tenían antecedentes de SIDA, el 20 % tenían un ARN del VIH-1 mayor de 100.000 copias por ml, el 13% recibieron ABC/3TC y el 87% recibieron FTC/TDF; estas características fueron similares entre los grupos de tratamiento. En el ensayo DRIVE-AHEAD, 728 pacientes fueron aleatorizados y recibieron al menos una dosis de doravirina/lamivudina/tenofovir disoproxilo 100/300/245 mg (DOR/3TC/TDF) o efavirenz/emtricitabina/tenofovir disoproxilo (EFV/FTC/TDF) 1 vez al día. En el momento basal, la mediana de edad de los pacientes era de 31 ańos (intervalo de 18 a 70 ańos), el 85% eran varones, el 52% eran de raza no blanca, el 3% tenían coinfección con hepatitis B o C, el 14% tenían antecedentes de SIDA, el 21% tenían una carga de ARN del VIH-1 > 100.000 copias por ml y el 12% tenían un recuento de linfocitos T CD4+ < 200 células/mm3; estas características fueron similares entre los grupos de tratamiento. En la Tabla 4 se presentan los resultados de los ensayos DRIVE-FORWARD y DRIVE-AHEAD en las semanas 48 y 96. Las pautas posológicas basadas en doravirina mostraron una eficacia uniforme entre todas las características demográficas y los factores pronósticos basales.
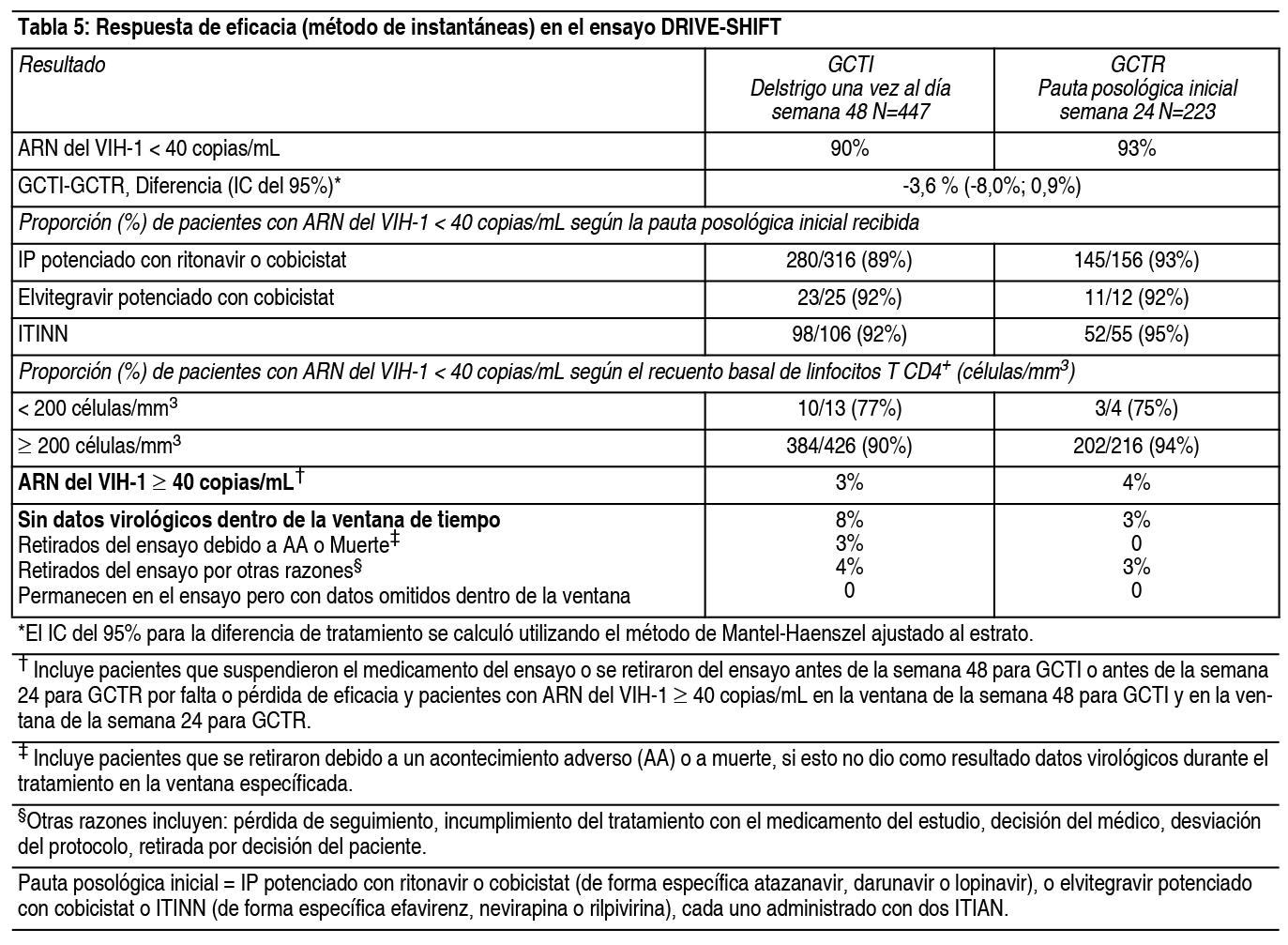
Pacientes adultos virológicamente suprimidos: La eficacia del cambio de tratamiento a Delstrigo desde una pauta posológica inicial que consiste en 2 inhibidores de la transcriptasa inversa análogos de nucleósido en combinación con un IP potenciado con ritonavir o cobicistat, o elvitegravir potenciado con cobicistat, o un ITIAN, se evaluó en un ensayo clínico abierto, aleatorizado (DRIVE-SHIFT) en adultos infectados por VIH-1 virológicamente suprimidos. Los pacientes debían haber sido virológicamente suprimidos (ARN del VIH-1 <40 copias/ml) con su pauta posológica inicial durante al menos 6 meses antes de la inclusión en el ensayo, sin antecedentes de fracaso virológico y ausencia documentada de sustituciones de RT que confieran resistencia a doravirina, lamivudina y tenofovir (ver la sección, Resistencia). Los pacientes fueron aleatorizados para cambiar el tratamiento a Delstrigo al inicio del ensayo [N=447, Grupo de Cambio de Tratamiento Inmediato (GCTI)] o permanecer con su pauta posológica inicial hasta la semana 24, momento en el cual se les cambió el tratamiento a Delstrigo [N=223, Grupo de Cambio de Tratamiento Retrasado (GCTR)]. Al inicio del ensayo, la mediana de la edad de los pacientes era de 43 ańos, 16% eran mujeres y 24% no eran de raza blanca. En el ensayo DRIVE-SHIFT, se vio que un cambio inmediato a Delstrigo mostraba no inferioridad en la semana 48 en comparación con la continuación de la pauta posológica inicial en la semana 24, según la proporción de pacientes con ARN del VIH-1 <40 copias/ml. Los resultados del tratamiento se muestran en la Tabla 5. Se observaron resultados coherentes para la comparación en la semana 24 del estudio en cada grupo de tratamiento.
Retiradas por acontecimientos adversos: En el ensayo DRIVE-AHEAD, la proporción de pacientes que abandonaron por un acontecimiento adverso en la semana 48 fue menor en el grupo de Delstrigo (3.0%) que en el grupo de EFV/FTC/TDF (6.6%). Población pediátrica: La Agencia Europea de Medicamentos ha concedido al titular un aplazamiento para presentar los resultados de los ensayos realizados con Delstrigo en 1 o más grupos de la población pediátrica en el tratamiento de la infección por el virus de la inmunodeficiencia humana de tipo 1 (VIH-1), en condición establecida en la decisión sobre los Planes de Investigación Pediátrico (PIP), para la indicación autorizada. Ver sección Posología para consultar la información sobre el uso en la población pediátrica. Propiedades farmacocinéticas: La administración de una dosis única de 1 comprimido de doravirina/lamivudina/tenofovir disoproxilo a voluntarios sanos (N=24) en ayunas deparó exposiciones similares de doravirina, lamivudina y tenofovir a la administración de comprimidos de doravirina (100 mg) más comprimidos de lamivudina (300 mg) más comprimidos de tenofovir disoproxilo (245 mg). La administración de un único comprimido de Delstrigo con una comida rica en grasas a voluntarios sanos produjo un aumento del 26% de la C24 de doravirina, mientras que el AUC y la Cmáx no se vieron afectados de manera significativa. La Cmáx de lamivudina disminuyó un 19% con una comida rica en grasas, mientras que el AUC no se vio afectado de manera significativa. La Cmáx de tenofovir disminuyó un 12% y el AUC aumentó un 27% con una comida rica en grasas. Estas diferencias farmacocinéticas no son clínicamente importantes. Doravirina: La farmacocinética de doravirina se estudió en voluntarios sanos y en pacientes infectados por el VIH-1. La farmacocinética de doravirina es similar en voluntarios sanos y en pacientes infectados por el VIH-1. El estado de equilibrio se alcanzó por norma general el día 2 de la administración 1 vez al día, con índices de acumulación de 1.2 a 1.4 para el AUC0-24, la Cmáx y la C24. A continuación, se presenta la farmacocinética en estado de equilibrio de doravirina tras la administración de 100 mg 1 vez al día a pacientes infectados por el VIH-1, de acuerdo con un análisis de farmacocinética poblacional.
Absorción: Tras la administración oral, las concentraciones plasmáticas máximas se alcanzan 2 horas después de la administración. Doravirina tiene una biodisponibilidad absoluta estimada aproximada del 64% para el comprimido de 100 mg. Distribución: De acuerdo a la administración de una microdosis IV, el volumen de distribución de doravirina es de 60.5 l. Doravirina se une de manera aproximada en un 76% a las proteínas plasmáticas. Biotransformación: Según datos obtenidos in vitro, doravirina se metaboliza de forma principal por CYP3A. Eliminación: Doravirina: Doravirina tiene una semivida terminal (t1/2) aproximada de 15 horas. Doravirina se elimina de manera principal a través del metabolismo oxidativo mediado por CYP3A4. La excreción biliar del medicamento inalterado puede contribuir a la eliminación de doravirina, pero no se espera que esta vía de eliminación sea significativa. La excreción urinaria del medicamento inalterado es secundaria. Lamivudina: Tras la administración oral, lamivudina se absorbe de forma rápida y se distribuye de manera amplia. Tras la administración oral de dosis múltiples de lamivudina 300 mg 1 vez al día durante 7 días a 60 voluntarios sanos, la Cmáx en estado de equilibrio (Cmáx,ee) fue de 2.04 ± 0.54 mcg por ml (media ± DE) y el AUC en estado de equilibrio durante 24 horas (AUC24,ee) fue de 8.87 ± 1.83 mcg•hora por ml. La unión a proteínas plasmáticas es baja. El 71% aproximado de una dosis intravenosa de lamivudina se recupera como medicamento inalterado en orina. El metabolismo de lamivudina es una vía de eliminación secundaria. En humanos, el único metabolito conocido es el metabolito trans-sulfóxido (el 5% aproximado de una dosis oral después de 12 horas). En la mayoría de los ensayos de dosis únicas en pacientes infectados por el VIH-1, o en voluntarios sanos con obtención de muestras de suero durante 24 horas después de la administración, la semivida de eliminación media observada (t˝) varió entre 5 y 7 horas. En pacientes infectados por el VIH-1, el aclaramiento total fue de 398.5 ± 69.1 por min (media ± DE). Tenofovir disoproxilo: Tras la administración oral de una dosis única de 245 mg de tenofovir disoproxilo a pacientes infectados por el VIH-1 en ayunas, la Cmáx se alcanzó en 1 hora. Los valores de Cmáx y AUC fueron de 0.30 ± 0.09 mcg por ml y 2.29 ± 0.9 ug.h por ml, de manera respectiva. La biodisponibilidad oral aproximada de tenofovir a partir de tenofovir disoproxilo en pacientes en ayunas es del 25%. Menos del 0.7% de tenofovir se une a proteínas plasmáticas humanas in vitro en el intervalo de 0.01 a 25 mcg por ml. De manera aproximada el 70-80% de la dosis intravenosa de tenofovir se recupera como medicamento inalterado en orina en las 72 horas siguientes a la administración. Tenofovir se elimina mediante una combinación de filtración glomerular y secreción tubular activa con un aclaramiento renal en adultos con un CrCl superior a 80 ml por minuto de 243.5 ± 33.3 ml por minuto (media ± DE). Tras la administración oral, la semivida terminal aproximada de tenofovir es de 12 a 18 horas. Estudios in vitro han determinado que ni tenofovir disoproxilo ni tenofovir son sustratos para las enzimas del CYP450. Insuficiencia renal: Doravirina: La excreción renal de doravirina es secundaria. En un estudio en el que se compararon 8 pacientes con insuficiencia renal grave y 8 pacientes sin insuficiencia renal, la exposición a una dosis única de doravirina fue un 31% mayor en los pacientes con insuficiencia renal grave. En un análisis de farmacocinética poblacional en el que participaron pacientes con CrCl entre 17 y 317 ml/min, la función renal no tuvo un efecto clínicamente importante sobre la farmacocinética de doravirina. No es necesario ajustar la dosis en los pacientes con insuficiencia renal leve, moderada o grave. La doravirina no se ha estudiado en pacientes con enfermedad renal terminal ni en pacientes sometidos a diálisis (ver sección Posología). Lamivudina: Los estudios con lamivudina muestran que las concentraciones plasmáticas (AUC) están aumentadas en pacientes con disfunción renal debido a una disminución del aclaramiento. De acuerdo con los datos de lamivudina, no se recomienda Delstrigo en pacientes con CrCl < 50 ml/min. Tenofovir disoproxilo: Los parámetros farmacocinéticos de tenofovir se determinaron tras la administración de una dosis única de 245 mg de tenofovir disoproxilo a 40 pacientes adultos no infectados por el VIH con distintos grados de insuficiencia renal, definida según el CrCl basal (función renal normal cuando el CrCl es > 80 ml/min; leve cuando el CrCl es de 50-79 ml/min; moderada cuando el CrCl es de 30-49 ml/min y grave cuando el CrCl es de 10-29 ml/min). En comparación con los pacientes con función renal normal, la exposición media (% CV) a tenofovir aumentó desde 2.185 (12%) ng·h/ml en los pacientes con CrCl > 80 ml/min hasta 3.064 (30%) ng·h/ml, 6.009 (42%) ng·h/ml y 15.985 (45%) ng·h/ml en los pacientes con insuficiencia renal leve, moderada y grave de forma respectiva. No se ha estudiado la farmacocinética de tenofovir en pacientes adultos no hemodializados con CrCl < 10 ml/min ni en pacientes con enfermedad renal terminal tratados con diálisis peritoneal u otras formas de diálisis. Insuficiencia hepática: Doravirina: Doravirina se metaboliza y elimina de manera principal por el hígado. En un estudio en el que se comparó a 8 pacientes con insuficiencia hepática moderada (clasificada como puntuación B de Child-Pugh sobre todo debido a un aumento de las puntuaciones de encefalopatía y ascitis) con 8 pacientes sin insuficiencia hepática, no se observaron diferencias clínicamente importantes en la farmacocinética de doravirina. No es necesario ajustar la dosis en pacientes con insuficiencia hepática leve o moderada. No se ha estudiado la doravirina en pacientes con insuficiencia hepática grave (clase C de Child-Pugh) (ver sección Posología). Lamivudina: Las propiedades farmacocinéticas de lamivudina se han determinado en pacientes con insuficiencia hepática moderada a grave. Los parámetros farmacocinéticos no se alteraron al disminuir la función hepática. No se ha establecido la seguridad y eficacia de lamivudina en presencia de hepatopatía descompensada. Tenofovir disoproxilo: Se ha estudiado la farmacocinética de tenofovir después de una dosis de 245 mg de tenofovir disoproxilo en voluntarios sanos con insuficiencia hepática moderada a grave. No se observaron diferencias clínicamente importantes en la farmacocinética de tenofovir entre pacientes con insuficiencia hepática y voluntarios sanos. Pacientes de edad avanzada: Aunque se incluyó un número limitado de pacientes a partir de 65 ańos (n=36), no se han encontrado diferencias clínicamente importantes en la farmacocinética de doravirina en pacientes a partir de 65 ańos en comparación con pacientes menores de 65 ańos en un ensayo de fase 1, ni en un análisis de farmacocinética poblacional. No se ha estudiado la farmacocinética de lamivudina y tenofovir en pacientes mayores de 65 ańos. No es necesario ajustar la dosis. Sexo: No se han identificado diferencias clínicamente importantes en la farmacocinética de doravirina, lamivudina y tenofovir entre hombres y mujeres. Raza: Doravirina: No se han identificado diferencias clínicamente importantes en relación a la raza en la farmacocinética de doravirina, según un análisis de farmacocinética poblacional de doravirina en voluntarios sanos y pacientes infectados por el VIH-1. Lamivudina: No existen diferencias raciales significativas ni clínicamente importantes en la farmacocinética de lamivudina. Tenofovir disoproxilo: No hubo números suficientes de grupos raciales y étnicos distintos de la raza blanca para determinar de manera adecuada las posibles diferencias farmacocinéticas entre estas poblaciones tras la administración de tenofovir disoproxilo. Datos preclínicos sobre seguridad: Toxicidad para la reproducción: Doravirina: Se han realizado estudios de reproducción con doravirina administrada por vía oral en ratas y conejos con exposiciones aproximadas de 9 veces (ratas) y 8 veces (conejos) la exposición en humanos a la dosis humana recomendada (DHR), sin efectos en el desarrollo embriofetal (ratas y conejos) o pre/posnatal (ratas). Los estudios realizados en ratas y conejas embarazadas demostraron que doravirina se transfiere al feto a través de la placenta, con concentraciones plasmáticas fetales de hasta el 40% (conejos) y el 52% (ratas) de las concentraciones maternas observadas el día 20 de gestación. Doravirina se excretó en la leche de ratas lactantes tras su administración oral, con concentraciones en la leche aproximadamente 1.5 veces mayores que las concentraciones plasmáticas de la madre. Lamivudina: Lamivudina no fue teratógena en estudios realizados con animales, pero hubo indicios de un aumento de las muertes embrionarias prematuras en conejos con exposiciones sistémicas relativamente bajas, comparables a las alcanzadas en humanos. No se observó un efecto similar en ratas ni siquiera con una exposición sistémica muy alta. Tenofovir disoproxilo: Los estudios de toxicidad para la reproducción en ratas y conejos no mostraron efectos sobre el apareamiento, la fertilidad, el embarazo ni los parámetros fetales. Sin embargo, tenofovir disoproxilo redujo el índice de viabilidad y el peso de las crías en un estudio de toxicidad peri-postnatal con dosis tóxicas para la madre. Carcinogenia: Doravirina: Los estudios de carcinogenicidad oral a largo plazo de doravirina en ratones y ratas no mostraron evidencias de potencial carcinogénico con exposiciones estimadas de hasta 6 veces (ratones) y 7 veces (ratas) las exposiciones humanas a la DHR. Lamivudina: Los estudios de carcinogenicidad a largo plazo de lamivudina en ratones y ratas no mostraron indicios de potencial carcinogénico con exposiciones de hasta 12 veces (ratones) y 57 veces (ratas) la exposición humana con la DHR. Tenofovir disoproxilo: Los estudios de carcinogenicidad oral en ratas y ratones solo revelaron una baja incidencia de tumores duodenales con dosis extremadamente altas en ratones. Es improbable que estos tumores sean relevantes para los humanos. Mutagénesis: Doravirina: Doravirina no fue genotóxica en una batería de pruebas in vitro o in vivo. Lamivudina: Lamivudina fue mutágena en una prueba de linfoma de ratón L5178Y y clastógena en un análisis citogenético con linfocitos humanos cultivados. Lamivudina no fue mutágena en una prueba de mutagenicidad microbiana, en una prueba de transformación celular in vitro, en una prueba de micronúcleos en ratas, en un análisis citogenético en médula ósea de rata y en un análisis de síntesis de ADN no programada en hígado de rata. Tenofovir disoproxilo: Tenofovir disoproxilo fue mutágeno en la prueba de linfoma de ratón in vitro y negativo en la prueba de mutagenicidad microbiana in vitro (prueba de Ames). En una prueba de micronúcleos de ratón in vivo, tenofovir disoproxilo fue negativo cuando se administró a ratones macho. Deterioro de la fertilidad: Doravirina: No se observaron efectos sobre la fertilidad, el rendimiento del apareamiento ni el desarrollo embrionario inicial cuando se administró doravirina a ratas hasta 7 veces la exposición en seres humanos a la DHR. Lamivudina: Lamivudina no afectó a la fertilidad masculina o femenina en ratas. Tenofovir disoproxilo: Los estudios de toxicidad para la reproducción en ratas y conejos no mostraron efectos sobre el apareamiento, la fertilidad, el embarazo ni los parámetros fetales. Toxicidad por dosis repetidas: Doravirina: La administración de doravirina en estudios de toxicidad en animales no se asoció a toxicidad. Lamivudina: La administración de lamivudina en estudios de toxicidad en animales a dosis altas no se asoció a toxicidad orgánica importante. En los niveles de dosificación más altos, se observaron efectos poco importantes en los indicadores de la función hepática y renal junto con reducciones ocasionales en el peso del hígado. Los efectos clínicamente relevantes observados fueron una reducción en el recuento de glóbulos rojos y neutropenia. Tenofovir disoproxilo: Los resultados de los estudios de toxicidad de dosis repetidas en ratas, perros y monos con niveles de exposición iguales o superiores a los niveles de exposición clínica y con posible relevancia para el uso clínico incluyeron cambios renales y óseos y una disminución de la concentración sérica de fosfato. La toxicidad ósea se diagnosticó como osteomalacia (monos) y reducción de la densidad mineral ósea (DMO) (ratas y perros). La toxicidad ósea en ratas y perros adultos jóvenes ocurrió a exposiciones ≥ 5 veces la exposición en pacientes pediátricos o adultos; la toxicidad ósea se produjo en monos infectados jóvenes a exposiciones muy altas después de la administración subcutánea (≥ 40 veces la exposición en pacientes). Los hallazgos en los estudios con ratas y monos indicaron que hubo una disminución relacionada con la sustancia en la absorción intestinal de fosfato con una posible reducción secundaria en la DMO.
Posología:Posología y forma de administración: El tratamiento debe ser iniciado por un médico con experiencia en el tratamiento de la infección por el VIH. Posología: La dosis recomendada de Delstrigo 100/300/245 mg es de 1 comprimido administrado por vía oral 1 vez al día, con o sin alimentos. Ajuste de la dosis: Si se administra Delstrigo junto con rifabutina, se debe aumentar la dosis de doravirina a 100 mg 2 veces al día. Esta dosis se alcanza ańadiendo 1 comprimido de doravirina de 100 mg (como fármaco único), para tomarse aproximadamente con 12 horas de diferencia de la dosis de Delstrigo (ver sección Interacción con otros medicamentos y otras formas de interacción). No se ha evaluado la administración de doravirina junto con otros inductores moderados de CYP3A, pero se esperan disminuciones en las concentraciones de doravirina. Si no se puede evitar la administración junto con otros inductores moderados de CYP3A (p.ej., dabrafenib, lesinurad, bosentán, tioridazina, nafcilina, modafinilo, telotristat de etilo), se debe tomar 1 comprimido diario de 100 mg de doravirina, aproximadamente 12 horas después de la dosis de Delstrigo (ver sección Interacción con otros productos medicinales y otras formas de interacción). No se ha evaluado la administración de doravirina junto con otros inductores moderados de CYP3A, pero se esperan disminuciones en las concentraciones de doravirina. Si no se puede evitar la administración junto con otros inductores moderados de CYP3A (p. ej. dabrafenib, lesinurad, bosentán, tioridazina, nafcilina, modafinilo, telotristat de etilo), se debe tomar 1 comprimido diario de 100 mg de doravirina, aproximadamente 12 horas después de la dosis de Delstrigo (ver sección Interacción con otros productos medicinales y otras formas de interacción). Dosis olvidadas: Si el paciente olvida una dosis de Delstrigo y han transcurrido menos de 12 horas desde la última dosis programada, deberá tomar Delstrigo lo antes posible y proseguir con el tratamiento de la manera habitual. Si el paciente olvida una dosis de Delstrigo y han transcurrido más de 12 horas desde la última dosis programada, no deberá tomar la dosis olvidada y en su lugar esperará a tomar la siguiente dosis a la hora programada de manera habitual. El paciente no debe tomar 2 dosis al mismo tiempo. Poblaciones especiales: Pacientes de edad avanzada: Se dispone de datos limitados sobre el uso de doravirina, lamivudina y tenofovir disoproxilo en pacientes a partir de 65 ańos. No hay evidencia de que los pacientes de edad avanzada necesiten una dosis diferente a la de los pacientes adultos más jóvenes (ver sección Propiedades farmacocinéticas). Se aconseja especial atención en este grupo de edad debido a los cambios asociados a la edad, como disminución de la función renal (ver sección Advertencias y precauciones especiales de empleo). Insuficiencia renal: No es necesario ajustar la dosis de Delstrigo en adultos con un aclaramiento de creatinina (CrCl) estimado ≥50 ml/min. El tratamiento con Delstrigo no se debe iniciar en pacientes con un CrCl estimado <50 ml/min (ver las secciones Advertencias especiales y precauciones especiales para el uso y Propiedades farmacocinéticas). Insuficiencia hepática: No es necesario ajustar la dosis de doravirina/lamivudina/tenofovir disoproxilo en pacientes con insuficiencia hepática leve (clase A de Child-Pugh) o moderada (clase B de Child-Pugh). No se ha estudiado el uso de doravirina en pacientes con insuficiencia hepática grave (clase C de Child-Pugh). Se desconoce si la exposición a doravirina aumentará en pacientes con insuficiencia hepática grave. Por lo tanto, se aconseja tener precaución cuando se administre doravirina/lamivudina/tenofovir disoproxilo a pacientes con insuficiencia hepática grave (ver sección Propiedades farmacocinéticas). Población pediátrica: No se han establecido la seguridad y la eficacia de Delstrigo en pacientes menores de 18 ańos. No se dispone de datos. Forma de administración: Delstrigo se debe tomar por vía oral, 1 vez al día con o sin alimentos y se debe tragar entero (ver sección Propiedades farmacocinéticas).
Modo de Empleo:Precauciones especiales de eliminación y otras manipulaciones: La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
Efectos Colaterales:Resumen del perfil de seguridad: Las reacciones adversas notificadas con más frecuencia que se consideraron posibles o probables relacionadas con doravirina fueron náuseas (4%) y cefalea (3%). Tabla de reacciones adversas: Las reacciones adversas con relación supuesta (o al menos posible) con el tratamiento se enumeran a continuación por sistema de clasificación de órganos y frecuencia. Las reacciones adversas se enumeran en orden decreciente de gravedad dentro de cada intervalo de frecuencia. Las frecuencias se definen como muy frecuentes (≥1/10), frecuentes (≥ 1/100 a < 1/10), poco frecuentes (≥1/1.000 a <1/100), raras (≥1/10.000 a < 1/1.000) o muy raras (< 1/10.000). Tabla 2: Tabla de reacciones adversas asociadas con doravirina/lamivudina/tenofovir disoproxilo. Frecuencia: Reacciones adversas. Trastornos de la sangre y del sistema linfático: Poco frecuentes: neutropenia*, anemia*, trombocitopenia*. Muy raras: aplasia eritrocitaria pura*. Infecciones e infestaciones: Raras: Erupción pustular. Trastornos del metabolismo y de la nutrición: Poco frecuentes: hipofosfatemia, hipocaliemia*. Raras: hipomagnesemia, acidosis láctica*. Trastornos psiquiátricos: Frecuentes: sueńos anormales, insomnio1. Poco frecuentes: pesadilla, depresión2, ansiedad3, irritabilidad, estado confusional, ideación suicida. Raras: agresión, alucinación, trastorno de adaptación, alteración del humor, sonambulismo. Trastornos del sistema nervioso: Frecuentes: cefalea, mareo, somnolencia. Poco frecuentes: alteración de la atención, deterioro de la memoria, parestesia, hipertonía, sueńo deficiente. Muy raras: neuropatía periférica (o parestesia)*. Trastornos vasculares: Poco frecuentes: hipertensión. Trastornos respiratorios, torácicos y mediastínicos: Frecuentes: tos*, síntomas nasales*. Raras: disnea, hipertrofia de amígdalas. Trastornos gastrointestinales: Frecuentes: náuseas, diarrea, dolor abdominal4, vómitos, flatulencia. Poco frecuentes: estreńimiento, molestia abdominal5, distensión abdominal, dispepsia, heces blandas6, trastorno de la motilidad gastrointestinal7, pancreatitis*. Raras: tenesmo rectal. Trastornos hepatobiliares: Raras: esteatosis hepática*, hepatitis*. Trastornos de la piel y del tejido subcutáneo: Frecuentes: alopecia*, erupción8. Poco frecuentes: prurito. Raras: dermatitis alérgica, rosácea, angioedema*. Trastornos musculoesquéleticos y del tejido conjuntivo: Frecuentes: trastornos musculares*. Poco frecuentes: mialgia, artralgia, rabdomiólisis*+, debilidad muscular*+. Raras: dolor musculoesquelético, osteomalacia (se manifiesta como dolor óseo y rara vez contribuye a fracturas)*, miopatía*. Trastornos renales y urinarios: Poco frecuentes: creatinina elevada*, tubulopatía renal proximal (incluido el síndrome de Fanconi)*. Raras: lesión renal aguda, trastorno renal, cálculo urinario, nefrolitiasis, lesión renal aguda*, trastorno renal*, necrosis tubular aguda*, nefritis (incluida nefritis intersticial aguda)*, diabetes insípida nefrogénica*. Trastornos generales y alteraciones en el lugar de administración: Frecuentes: fatiga, fiebre*. Poco frecuentes: astenia, malestar general. Raras: dolor torácico, escalofríos, dolor, sed. Exploraciones complementarias: Frecuentes: alanina aminotransferasa elevada9. Poco frecuentes: aspartato aminotransferasa elevada, lipasa elevada, amilasa elevada, hemoglobina disminuida. Raras: creatinfosfoquinasa en sangre elevada. *Esta reacción adversa no se identificó como una reacción adversa asociada con doravirina a partir de los estudios clínicos de fase 3 (DRIVE-FORWARD, DRIVE-AHEAD, DRIVE-SHIFT), pero está incluida en esta tabla como una reacción adversa descrita en la Ficha Técnica de 3TC y/o TDF. Se utilizó la categoría de frecuencia más alta notificada en la Ficha Técnica de 3TC o TDF. +Esta reacción adversa se puede producir como consecuencia de tubulopatía renal proximal. En ausencia de esta enfermedad, no se considera causalmente asociado con tenofovir disoproxilo. 1insomnio incluye: insomnio, insomnio inicial y trastorno del sueńo. 2depresión incluye: depresión, estado de ánimo deprimido, depresión mayor y trastorno depresivo persistente. 3ansiedad incluye: ansiedad y trastorno de ansiedad generalizada. 4dolor abdominal incluye: dolor abdominal y dolor en la zona superior del abdomen. 5molestia abdominal incluye: molestia abdominal y malestar epigástrico. 6heces blandas incluye: heces blandas y heces anormales. 7trastorno de la motilidad gastrointestinal incluye: trastorno de la motilidad gastrointestinal y movimientos intestinales frecuentes. 8erupción incluye: erupción, erupción macular, erupción eritematosa, erupción generalizada, erupción maculopapular, erupción papular y urticaria. 9alanina aminotransferasa elevada incluye: alanina aminotransferasa elevada y lesión traumática hepatocelular. Notificación de sospechas de reacciones adversas: Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación.
Contraindicaciones: Hipersensibilidad a los principios activos o a alguno de los excipientes incluidos en la sección Lista de excipientes. Está contraindicada la administración junto con medicamentos que sean inductores potentes de las enzimas del citocromo P450 CYP3A, ya que se espera que se produzcan descensos significativos de las concentraciones plasmáticas de doravirina, lo que podría reducir la eficacia de Delstrigo (ver las secciones Advertencias y precauciones especiales de empleo e Interacciones medicamentosas y otras formas de interacción). Estos medicamentos son, entre otros, los siguientes: carbamazepina, oxcarbazepina, fenobarbital, fenitoína. Rifampicina, rifapentina. Hierba de San Juan (Hypericum perforatum). Mitotano. Enzalutamida. Lumacaftor.
Advertencias:Advertencias y precauciones especiales de empleo: Aunque se ha demostrado que una supresión viral eficaz con tratamiento antirretroviral reduce considerablemente el riesgo de transmisión sexual del VIH-1, no se puede descartar un riesgo residual. Se deben tomar precauciones para evitar la transmisión de acuerdo con las directrices nacionales. Sustituciones de ITINN y uso de doravirina: No se ha evaluado el uso de doravirina en pacientes con fracaso virológico previo a cualquier otro tratamiento antirretroviral. Las mutaciones asociadas a ITINN detectadas en la selección formaron parte de los criterios de exclusión en los estudios de fase 2b/3. No se ha establecido un punto de inflexión de la disminución de la sensibilidad, obtenida con distintas sustituciones de ITINN, que se asocie a una disminución de la eficacia clínica (ver sección Propiedades farmacodinámicas). No hay suficiente evidencia clínica para confirmar el uso de doravirina en pacientes infectados con el VIH-1 con evidencia de resistencia a ITINNs. Exacerbación aguda grave de la hepatitis B en pacientes coinfectados por el VIH-1 y el VHB: En todos los pacientes infectados por el VIH-1 se debe analizar la presencia del virus de la hepatitis B (VHB) antes de iniciar el tratamiento antirretroviral. Se han notificado exacerbaciones agudas graves de la hepatitis B (p. ej., con descompensación hepática e insuficiencia hepática) en pacientes coinfectados por el VIH-1 y el VHB, y que han interrumpido el tratamiento con lamivudina o tenofovir disoproxilo, dos de los componentes de Delstrigo. Los pacientes coinfectados por el VIH-1 y el VHB deben tener una vigilancia estrecha con seguimiento tanto clínico como analítico durante al menos varios meses después de interrumpir el tratamiento con Delstrigo. Si procede, puede estar justificado el inicio de un tratamiento contra la hepatitis B, de manera especial en pacientes con hepatopatía avanzada o cirrosis, ya que la exacerbación de la hepatitis después del tratamiento puede provocar descompensación hepática e insuficiencia hepática. Insuficiencia renal de nueva aparición o agravamiento de la insuficiencia renal ya existente: Se ha notificado insuficiencia renal, incluidos casos de insuficiencia renal aguda y síndrome de Fanconi (lesión tubular renal con hipofosfatemia grave), con el uso de tenofovir disoproxilo, un componente de Delstrigo. Se debe evitar el tratamiento con Delstrigo con el uso simultáneo o reciente de medicamentos nefrotóxicos (p. ej., antiinflamatorios no esteroideos [AINEs] en dosis altas o múltiples) (ver sección Interacciones con otros medicamentos y otras formas de interacción). Se han notificado casos de insuficiencia renal aguda tras iniciar tratamiento con AINEs a dosis altas o múltiples en pacientes infectados por el VIH con factores de riesgo de disfunción renal que parecían estables con tenofovir disoproxilo. Algunos pacientes precisaron hospitalización y tratamiento de sustitución renal. Se deben considerar alternativas a los AINEs, si es necesario, en los pacientes con riesgo de disfunción renal. La presencia de dolor óseo persistente o agravado, dolor en las extremidades, fracturas y/o dolor o debilidad muscular pueden ser manifestaciones de tubulopatía renal proximal y deben incitar a una evaluación de la función renal en los pacientes de riesgo. Se recomienda evaluar el CrCl estimado en todos los pacientes antes de iniciar el tratamiento y según proceda clínicamente durante el tratamiento con Delstrigo. En pacientes con riesgo de disfunción renal, incluidos los que hayan experimentado acontecimientos renales con anterioridad mientras recibían adefovir dipivoxilo, se recomienda evaluar el CrCl estimado, el fósforo sérico, la glucosa en orina y las proteínas en orina antes de iniciar el tratamiento con Delstrigo y realizar controles más frecuentes de la función renal según proceda, dependiendo de la situación médica del paciente durante el tratamiento con Delstrigo. Lamivudina y tenofovir disoproxilo se excretan de forma principal por el rińón. Se debe interrumpir el tratamiento con Delstrigo si el CrCl estimado desciende por debajo de 50 ml por minuto, ya que no se podrá lograr el ajuste necesario del intervalo de dosis de lamivudina y tenofovir disoproxilo con el comprimido de combinación de dosis fijas (ver sección Posología). Pérdida de hueso y defectos de la mineralización: Densidad mineral ósea: En ensayos clínicos realizados en adultos infectados por VIH-1, tenofovir disoproxilo se asoció con reducciones ligeramente mayores de la densidad mineral ósea (DMO) y aumentos de los marcadores bioquímicos del metabolismo óseo, lo que sugiere un aumento del recambio óseo con respecto a los medicamentos de comparación. Las concentraciones séricas de hormona paratiroidea y de 1.25 vitamina D fueron también mayores en los pacientes que recibieron tenofovir disoproxilo. En otros estudios (prospectivos y transversales), las reducciones más pronunciadas de la DMO se observaron en pacientes tratados con tenofovir disoproxilo como parte de un tratamiento que contenía un inhibidor de la proteasa potenciado. Las anomalías óseas (que rara vez contribuyen a fracturas) se pueden asociar a tubulopatía renal proximal. Se desconocen los efectos de los cambios asociados a tenofovir disoproxilo en la DMO y los marcadores bioquímicos en la salud ósea a largo plazo y el futuro riesgo de fracturas. Se debe considerar la evaluación de la DMO en los pacientes adultos infectados por el VIH-1 con antecedentes de fracturas óseas patológicas u otros factores de riesgo de osteoporosis o pérdida de hueso. Aunque no se ha estudiado el efecto de los suplementos de calcio y vitamina D, este tipo de suplementos podrían ser beneficiosos en todos los pacientes. Si se sospechan anomalías óseas, se deberá realizar una exploración apropiada. Defectos de mineralización: Se han notificado casos de osteomalacia asociada a tubulopatía renal proximal, que se manifiesta como dolor óseo o dolor en las extremidades y que puede contribuir al riesgo de fracturas, en asociación con el uso de tenofovir disoproxilo. También se han notificado artralgias y dolor o debilidad muscular en casos de tubulopatía renal proximal. Se debe considerar la aparición de hipofosfatemia y osteomalacia secundarias a tubulopatía renal proximal en pacientes con riesgo de disfunción renal que presenten síntomas óseos o musculares persistentes o agravados durante el tratamiento con medicamentos que contengan tenofovir disoproxilo (ver sección Advertencias y precauciones especiales de empleo). Administración junto con otros antivirales: No se debe administrar doravirina/lamivudina/tenofovir disoproxilo junto con otros medicamentos que contengan lamivudina, ni con medicamentos que contengan tenofovir disoproxilo o tenofovir alafenamida, ni con adefovir dipivoxilo (ver sección Interacción con otros medicamentos y otras formas de interacción). No se debe administrar doravirina/lamivudina/tenofovir disoproxilo junto con doravirina a menos que sea necesario para ajustar la dosis (p. ej., con rifabutina) (ver las secciones Posología e Interacción con otros medicamentos y otras formas de interacción). Uso con inductores de CYP3A: Se debe tener precaución al prescribir doravirina con medicamentos que puedan reducir la exposición de doravirina (ver las secciones Contraindicaciones e Interacción con otros medicamentos y otras formas de interacción). Síndrome de reconstitución inmune: Se ha notificado síndrome de reconstitución inmune en pacientes tratados con tratamiento antirretroviral de combinación. Durante la fase inicial del tratamiento antirretroviral de combinación, los pacientes cuyo sistema inmunitario responde, pueden presentar una respuesta inflamatoria a infecciones oportunistas inactivas o residuales (como infección por Mycobacterium avium, Citomegalovirus, neumonía por Pneumocystis jirovecii o tuberculosis), que precise una evaluación más detallada y tratamiento. También se han notificado trastornos autoinmunes (como enfermedad de Graves, polimiositis y síndrome de Guillain-Barré) en el contexto de una reconstitución inmune; sin embargo, el tiempo hasta su aparición es más variable y puede ocurrir muchos meses después del inicio del tratamiento. Lactosa: Delstrigo contiene lactosa monohidrato. Los pacientes con intolerancia hereditaria a galactosa, deficiencia total de lactasa o problemas de absorción de glucosa o galactosa no deben tomar este medicamento.
Precauciones:Fertilidad, embarazo y lactancia: Embarazo: No hay datos o estos son limitados relativos al uso de doravirina en mujeres embarazadas. Existe un elevado número de datos en mujeres embarazadas (más de 3.000 embarazos desde el primer trimestre) que indican que tomar el componente activo individual lamivudina en combinación con otros antirretrovirales no produce malformaciones. Existen algunos datos en mujeres embarazadas (entre 300-1.000 embarazos) que indican que tenofovir disoproxilo no produce malformaciones ni toxicidad fetal/neonatal. Registro de embarazos de las pacientes tratadas con antirretrovirales: Para vigilar los resultados maternofetales en pacientes embarazadas expuestas a antirretrovirales durante el embarazo, se ha establecido un registro de embarazos de las pacientes tratadas con antirretrovirales. Se recomienda a los médicos que incluyan a las pacientes en este registro. Los estudios en animales con doravirina no sugieren efectos perjudiciales directos ni indirectos en términos de toxicidad para la reproducción (ver sección Propiedades farmacocinéticas). Los estudios realizados en animales con tenofovir disoproxilo no sugieren efectos perjudiciales directos ni indirectos en términos de toxicidad para la reproducción (ver sección Propiedades farmacocinéticas). Los estudios en animales con lamivudina mostraron un aumento de las muertes embrionarias precoces en conejos, pero no en ratas (ver sección Propiedades farmacocinéticas). Se ha demostrado la transferencia placentaria de lamivudina en seres humanos. Lamivudina puede inhibir la replicación del ADN celular (ver sección Propiedades farmacocinéticas). Se desconoce la trascendencia clínica de este hallazgo. Como medida de precaución, es preferible evitar el uso de Delstrigo durante el embarazo. Lactancia: Se desconoce si doravirina se excreta en la leche materna. Los datos farmacodinámicos/toxicológicos disponibles en animales muestran que doravirina se excreta en la leche (ver sección Propiedades farmacocinéticas). Se ha encontrado lamivudina en recién nacidos/lactantes de mujeres tratadas. Según se desprende de la información procedente de más de 200 parejas de madre/hijo tratadas contra el VIH, las concentraciones séricas de lamivudina en los lactantes de madres tratadas contra el VIH son muy bajas (<4 % de las concentraciones séricas maternas) y disminuyen de forma progresiva hasta niveles indetectables cuando los lactantes alcanzan las 24 semanas de edad. No se dispone de datos sobre la seguridad de lamivudina cuando se administra a bebés menores de 3 meses. Tenofovir se excreta en la leche materna. No hay datos suficientes sobre los efectos de tenofovir en recién nacidos/lactantes. Debido a la posibilidad de transmisión del VIH-1 y a la posibilidad de reacciones adversas graves en lactantes, se debe indicar a las madres que no den el pecho si están tomando Delstrigo. Fertilidad: No se dispone de datos en seres humanos sobre el efecto de Delstrigo en la fertilidad. Los estudios en animales no sugieren efectos perjudiciales de doravirina, lamivudina o tenofovir disoproxilo en la fertilidad con niveles de exposición superiores a la exposición en humanos a la dosis clínica recomendada (ver sección Propiedades farmacocinéticas). Efectos sobre la capacidad para conducir y utilizar máquinas: Delstrigo puede tener una pequeńa influencia sobre la capacidad para conducir o utilizar máquinas. Se debe informar a los pacientes que se ha notificado cansancio, mareo y somnolencia durante el tratamiento con Delstrigo (ver sección Efectos colaterales). Esto se debe tener en cuenta al evaluar la capacidad de un paciente para conducir o utilizar máquinas.
Interacciones Medicamentosas:Interacción con otros medicamentos y otras formas de interacción: Delstrigo es una pauta posológica completa para el tratamiento de la infección por el VIH-1; por consiguiente, Delstrigo no se debe administrar junto con otros antirretrovirales. No se facilita información sobre las posibles interacciones farmacológicas con otros antirretrovirales. Los estudios de interacciones se han realizado sólo en adultos. Delstrigo contiene doravirina, lamivudina y tenofovir disoproxilo, por lo que cualquier interacción conocida de estos medicamentos individualmente se aplica a Delstrigo y se presentan en la Tabla 1. Efectos de otros medicamentos sobre doravirina, lamivudina y tenofovir disoproxilo: Doravirina: Doravirina se metaboliza de manera principal por CYP3A y se espera que los medicamentos que inducen o inhiben CYP3A afecten al aclaramiento de doravirina (ver sección Propiedades farmacocinéticas). Doravirina/lamivudina/tenofovir disoproxilo no se debe administrar junto con medicamentos que sean inductores potentes de las enzimas CYP3A, ya que se espera que se produzcan descensos importantes de las concentraciones plasmáticas de doravirina, lo que podría reducir la eficacia de doravirina/lamivudina/tenofovir disoproxilo (ver las secciones Contraindicaciones y Propiedades farmacocinéticas). La administración junto con rifabutina, inductor moderado de CYP3A, redujo las concentraciones de doravirina (ver Tabla 1). Cuando se administre Delstrigo junto con rifabutina, se debe administrar una dosis diaria de 100 mg de doravirina, aproximadamente 12 horas después de la dosis de doravirina/lamivudina/tenofovir disoproxilo (ver sección Posología). No se ha evaluado la administración de doravirina/lamivudina/tenofovir disoproxilo junto con otros inductores moderados de CYP3A, pero se esperan disminuciones en las concentraciones de doravirina. Si no se puede evitar la administración junto con otros inductores moderados de CYP3A (p. ej., dabrafenib, lesinurad, bosentán, tioridazina, nafcilina, modafinilo, telotristat de etilo), se debe administrar una dosis diaria de 100 mg de doravirina, aproximadamente 12 horas después de la administración de doravirina/lamivudina/tenofovir disoproxilo (ver sección Posología). La administración de doravirina/lamivudina/tenofovir disoproxilo junto con medicamentos que son inhibidores de CYP3A puede aumentar las concentraciones plasmáticas de doravirina. Sin embargo, no es necesario ajustar la dosis de doravirina cuando se administre junto con inhibidores de CYP3A. Lamivudina: Dado que lamivudina se elimina de forma principal por los rińones mediante una combinación de filtración glomerular y secreción tubular activa (ver sección Propiedades farmacocinéticas), la administración de doravirina/lamivudina/tenofovir disoproxilo junto con medicamentos que reducen la función renal o compiten por la secreción tubular activa puede aumentar las concentraciones séricas de lamivudina. Tenofovir disoproxilo: Dado que tenofovir se elimina de forma principal por los rińones mediante una combinación de filtración glomerular y secreción tubular activa (ver la sección Propiedades farmacocinéticas), la administración de doravirina/lamivudina/tenofovir disoproxilo junto con medicamentos que reducen la función renal o compiten por la secreción tubular activa a través de OAT1, OAT3 o MRP4 puede aumentar las concentraciones séricas de tenofovir. Debido al componente tenofovir disoproxilo de la combinación doravirina/lamivudina/tenofovir disoproxilo, se debe evitar el uso simultáneo de este medicamento con medicamentos nefrotóxicos o que se hayan tomado de manera reciente. Algunos ejemplos son, entre otros, aciclovir, cidofovir, ganciclovir, valaciclovir, valganciclovir, aminoglucósidos (p. ej., gentamicina) y AINEs en dosis altas o múltiples (ver sección Advertencias especiales y precauciones especiales para el uso). Efectos de doravirina, lamivudina y tenofovir disoproxilo sobre otros medicamentos: Doravirina: No es probable que doravirina, en dosis de 100 mg 1 vez al día, tenga un efecto clínicamente importante en las concentraciones plasmáticas de medicamentos que dependan de proteínas transportadoras para su absorción y/o eliminación o que se metabolicen mediante enzimas CYP. Sin embargo, la administración de doravirina junto con midazolam, un sustrato de CYP3A sensible, dio lugar a una disminución del 18% en la exposición de midazolam, lo que indica que doravirina puede ser un inductor débil de CYP3A. Por lo tanto, se debe tener precaución cuando se administre doravirina junto con medicamentos que son sustratos de CYP3A sensibles que también tengan un margen terapéutico estrecho (p. ej., tacrolimus y sirolimus). Lamivudina: Lamivudina no inhibe ni induce las enzimas CYP. Tenofovir: De acuerdo con los resultados de experimentos in vitro y la vía de eliminación conocida de tenofovir, la posibilidad de interacciones mediadas por el CYP entre tenofovir y otros medicamentos es pequeńa. Tabla de interacciones: En la Tabla 1 se muestran las interacciones conocidas y otras interacciones posibles de medicamentos con los componentes individuales de Delstrigo, pero no están todas incluidas (el aumento se indica como ←, la disminución se indica cómo ↓ y la ausencia de cambios se indica como ↔). Para conocer las interacciones posibles de medicamentos con tenofovir disoproxilo o lamivudina, (ver las secciones Advertencias y precauciones especiales de empleo y Propiedades farmacocinéticas).
Sobredosificación:Doravirina: No existe información sobre posibles síntomas agudos y signos de sobredosis con doravirina. Lamivudina: Debido a que la cantidad de lamivudina eliminada mediante hemodiálisis (4 horas), diálisis peritoneal ambulatoria continua y diálisis peritoneal automatizada es insignificante, se desconoce si la hemodiálisis continua proporcionaría un beneficio clínico en un episodio de sobredosis de lamivudina. Tenofovir disoproxilo: Tenofovir disoproxilo se elimina de manera eficiente mediante hemodiálisis con un coeficiente de extracción aproximado del 54%. Después de una dosis única de 245 mg de tenofovir disoproxilo, una sesión de 4 horas de hemodiálisis permitió eliminar aproximadamente el 10% de la dosis administrada de tenofovir.
Incompatibilidades: No procede.
Conservación:Período de validez: 24 meses almacenado a no más de 25° C. Precauciones especiales de conservación: Conservar en el frasco original y mantener el frasco perfectamente cerrado para protegerlo de la humedad. No retirar el desecante. Este medicamento no requiere ninguna temperatura especial de conservación.
Presentaciones:Naturaleza y contenido del envase: Cada caja contiene 1 frasco de polietileno de alta densidad (HDPE) con cierre de polipropileno a prueba de nińos y desecante de gel de sílice. Están disponibles los siguientes tamańos de envase: 1 frasco conteniendo 30 comprimidos recubiertos.
 Laboratorio
Laboratorio