Composición: Cada comprimido recubierto contiene: Capecitabina 500 mg. Excipientes: Lactosa Anhidra, Croscarmelosa Sódica, Hipromelosa, Celulosa Microcristalina, Estearato de Magnesio, Dióxido de Titanio, Oxido de Hierro Rojo, Oxido de Hierro Amarillo, c.s.
Descripción: Xenevia 500 contiene capecitabina, principio activo que pertenece al grupo farmacoterapéutico de los citostáticos (antimetabolito). La capecitabina es un carbamato de fluoropirimidina no citotóxica que, administrado por vía oral, actúa como un precursor (pro-fármaco) del citotóxico 5-fluorouracil (5-FU). La capecitabina se activa a través de varios pasos enzimáticos.
Acción Terapéutica: Antineoplásico.
Indicaciones:Carcinoma de mama: En asociación con docetaxel, Xenevia 500 está indicado para el tratamiento del carcinoma de mama localmente avanzado o metastásico, tras el fracaso de la quimioterapia citotóxica o con una antraciclina. En monoterapia, Xenevia 500 está indicada para el tratamiento del carcinoma de mama localmente avanzado o metastásico cuando haya fracasado la quimioterapia con taxanos y antraciclinas o cuando no esté indicado proseguir el tratamiento con antraciclinas. Carcinoma colorrectal: Xenevia 500 está indicado como tratamiento adyuvante del cáncer de colon etapa III (etapa C de Dukes), en pacientes que han sufrido resección completa del tumor primario. Xenevia 500 está indicado para el tratamiento de primera línea del cáncer colorrectal metastásico como monoterapia o en combinación con oxaliplatino, con o sin bevacizumab. Carcinoma esofagogástrico: Xenevia 500 está indicado como tratamiento de primera línea del cáncer esofagogástrico avanzado o metastásico, en asociación con epirrubicina y oxaliplatino o cisplatino. Cáncer gástrico: En combinación con oxaliplatino, Xenevia 500 está indicado como tratamiento adyuvante de pacientes luego de la resección completa de adenocarcinoma gástrico en etapa II y III.
Propiedades:Farmacología clínica: Mecanismo de acción: La capecitabina es un carbamato de fluoropirimidina no citotóxica que, administrado por vía oral, actúa como un precursor del citotóxico 5-fluorouracil (5-FU). La capecitabina se activa a través de varios pasos enzimáticos. La enzima responsable de la conversión final a 5-FU, la timidina fosforilasa (ThyPasa), se encuentra en tejidos tumorales así como en tejidos normales aunque con niveles generalmente más bajos. Los metabolitos del 5-FU causan dańo celular por 2 mecanismos. En primer término, el 5-fluoro-2' deoxyuridine monophosphate (FdUMP) y el cofactor de folato (N5-10- methylenetetrahydrofolate) se une a la timidilata sintetasa (TS), bloqueando la reacción de metilación del ácido desoxiuridílico hacia el ácido timidílico, por ello interfiere con la síntesis del ácido desoxirribonucleico (ADN), de manera que una deficiencia de este compuesto puede inhibir la división celular. En segundo término, las enzimas de la transcripción nuclear pueden incorporar erróneamente 5-fluorouridine triphosphate (FUTP) en lugar de la uridina trifosfato (UTP) durante la síntesis del ARN, este error metabólico puede interferir con el procesamiento del ARN y la síntesis proteica. Los efectos de la privación del ADN y el ARN se acentúan en las células que proliferan más rápidamente y que metabolizan 5-FU con mayor velocidad. Propiedades farmacocinéticas: Absorción: Tras la administración oral de 1250 mg/m2 2 veces al día a pacientes con cáncer, la capecitabina atraviesa la mucosa intestinal en forma de molécula intacta y se absorbe de modo rápido y extenso, transformándose posteriormente de forma amplia en los metabolitos 5'-DFCR y 5'-DFUR. Capecitabina alcanza las concentraciones plasmáticas máximas (Tmax) en unas 1.5 horas con un pico posterior del 5-FU a las 2 horas. La administración con los alimentos reduce la velocidad y la tasa de absorción de la capecitabina, modifica el valor de las concentraciones plasmáticas máximas (Cmax) y el ABC con una reducción media de un 60 y 35% respectivamente. La Cmax y el ABC0-∞ del 5-FU también se redujeron por la comida un 43% y 21% respectivamente. La comida retrasó el tiempo para las concentraciones plasmáticas máximas (Tmax en horas) a 1.50 para la capecitabina y el 5-FU. Distribución: La unión a proteínas plasmáticas, sobre todo a la albúmina, de la capecitabina y sus metabolitos (5'-DFCR, el 5'-DFUR y el 5-FU) fue menor al 60% (en un 54%, 10%, 62% y 10%, respectivamente). La capecitabina tiene un bajo potencial para interacciones farmacocinéticas relacionadas a la unión a proteínas plasmáticas. Biotransformación y metabolismo: La capecitabina es extensamente metabolizada a 5-FU. En el hígado la carboxilesterasa hidroliza a la mayoría del compuesto 5'-DFCR. La citidina deaminasa, una enzima presente en la mayoría de los tejidos incluso los tumores, convierte subsecuentemente al 5'-DFCR en 5-FU. La enzima timidina fosforilasa (dThyPase) entonces hidroliza al 5'-DFUR a el fármaco activo 5-FU. Las enzimas que intervienen en la activación catalítica se localizan en los tejidos tumorales, pero también se encuentran en los tejidos sanos, pero normalmente en niveles más bajos. Después de la administración oral de capecitabina a pacientes con cáncer colorrectal, la relación entre concentración de 5-FU en los tumores colorrectales y los tejidos adyacentes fue 2.9 (oscilo de 0.9 a 8.0). Estas relaciones no han sido evaluadas en pacientes con cáncer de mama o comparado con la infusión de 5-FU. Posteriormente la enzima dihidropirimidin dehidrogenasa (DPD), hidrogena al 5-FU el producto del metabolismo de capecitabina, a uno mucho menos tóxico el 5-fluoro-5,6 dihidro-fluorouracil (FUH2). La dihidropirimidinasa rompe el anillo de pirimidina y produce el ácido 5-fluoro-ureidopropionico (FUPA). Finalmente, la β-ureido-propionasa transforma el FUPA a αfluoro-β-alanina (FBAL) la cual es eliminada por la orina. También se ha determinado que capecitabina y sus metabolitos (5'-DFCR, 5'-DFUR, 5-FU y FBAL) no inhiben el metabolismo de los sustratos de prueba por las isoenzimas de citocromo P450: 1A2, 2A6, 3A4, 2C19, 2D6 y 2E1. Eliminación: La capecitabina y sus metabolitos se eliminan fundamentalmente por excreción urinaria. El 95.5% de la dosis administrada de capecitabina se recoge en orina. La excreción fecal es mínima (2.6 %). El principal metabolito excretado en la orina es FBAL, representando un 57% de la dosis administrada. Alrededor del 3% de la dosis administrada se excreta inalterada por la orina. La vida media de eliminación (t1/2 en horas) de capecitabina, y 5-FU fue 0.85 y, 0.76 respectivamente. Terapia en combinación: Los ensayos fase I para evaluar el efecto de capecitabina sobre la farmacocinética de docetaxel o paclitaxel y viceversa mostraron que no existe efecto de capecitabina sobre la farmacocinética de docetaxel o paclitaxel (Cmax y AUC) ni del docetaxel o paclitaxel sobre la farmacocinética del 5'-DFUR. Farmacocinética en poblaciones especiales: Se han realizado análisis de farmacocinética en la población tras el tratamiento con capecitabina en pacientes con cáncer colorrectal a dosis de 1250 mg/m2 2 veces al día, que indicaron que el sexo, la raza, la edad, la presencia o ausencia de metástasis hepáticas basales, no tuvieron un efecto estadísticamente significativo sobre la farmacocinética del 5'-DFUR, 5-FU y FBAL. Pacientes con insuficiencia hepática debida a metástasis hepática: Según evaluaciones farmacocinéticas realizadas en pacientes cancerosos con insuficiencia hepática leve a moderada causada por metástasis hepáticas, la biodisponibilidad de capecitabina y la exposición a 5-FU puede aumentarse (tanto la Cmax y el ABC0-∞se incrementaron un 60%) si se compara con pacientes sin insuficiencia hepática. Por lo que se deberá ser cuidadoso al administrar capecitabina en este tipo de paciente. No se disponen de datos farmacocinéticas en pacientes con insuficiencia hepática grave (ver en Precauciones y advertencias). Pacientes con insuficiencia renal: En base a evaluaciones de farmacocinética en pacientes cancerosos con insuficiencia renal leve a grave, tratados con capecitabina, se observó que el aclaramiento de creatinina tiene influencia sobre la exposición sistémica a 5' DFUR (42% de incremento en el AUC) y a FBAL (85% de aumento del AUC) cuando existe una insuficiencia renal moderada. Estas cifras aumentan a un 258% y 71% respectivamente cuando se usa el fármaco en insuficiencia renal severa. FBAL es un metabolito sin actividad antiproliferativa. La exposición sistémica de capecitabina se incrementa un 25% tanto en pacientes con insuficiencia renal moderada como severa (ver Posología, Precauciones y Advertencias, uso en poblaciones especiales). Efecto de capecitabina sobre la farmacocinética de warfarina: La administración crónica de capecitabina a pacientes con cáncer con una dosis única de 20 mg de warfarina aumentó el ABC de S-warfarina en un 57% y disminuyó su aclaramiento en un 37%, la línea basal del ABC corregido del JNR en estos pacientes aumentó en 2.8 veces, y el máximo valor medio observado la lNR se incrementó en un 91% (ver Interacciones con otros medicamentos). Efecto de los antiácidos sobre la farmacocinética de capecitabina: Cuando se administró un antiácido como hidróxido de aluminio e hidróxido de magnesio, inmediatamente después de capecitabina en pacientes con cáncer, el ABC y la Cmax aumentaron en un 16% y 35%, respectivamente, para capecitabina y en un 18% y 22%, respectivamente, para 5'-DFCR. No se observó efecto en los otros 3 principales metabolitos de capecitabina (5'-DFUR, 5-FU, FBAL). Efecto de capecitabina sobre la farmacocinética de docetaxel y viceversa: Se evaluó el efecto de capecitabina sobre la farmacocinética de docetaxel y el efecto de docetaxel en la farmacocinética de capecitabina en pacientes con tumores sólidos. La capecitabina mostró que no tiene efecto sobre la farmacocinética de docetaxel (Cmax y AUC) y docetaxel no tiene ningún efecto sobre la farmacocinética de la capecitabina y el precursor de 5-FU 5' DFUR.
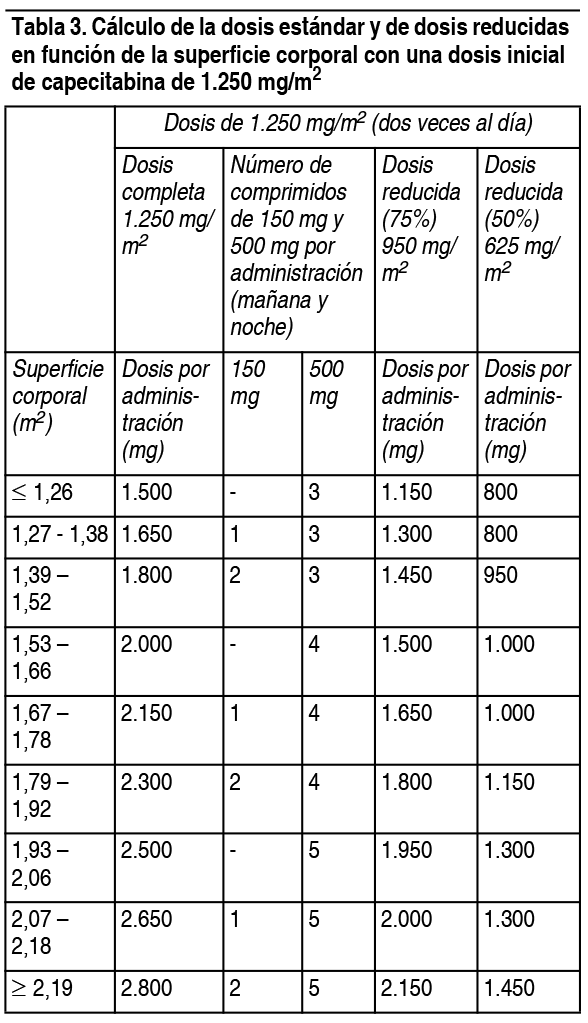
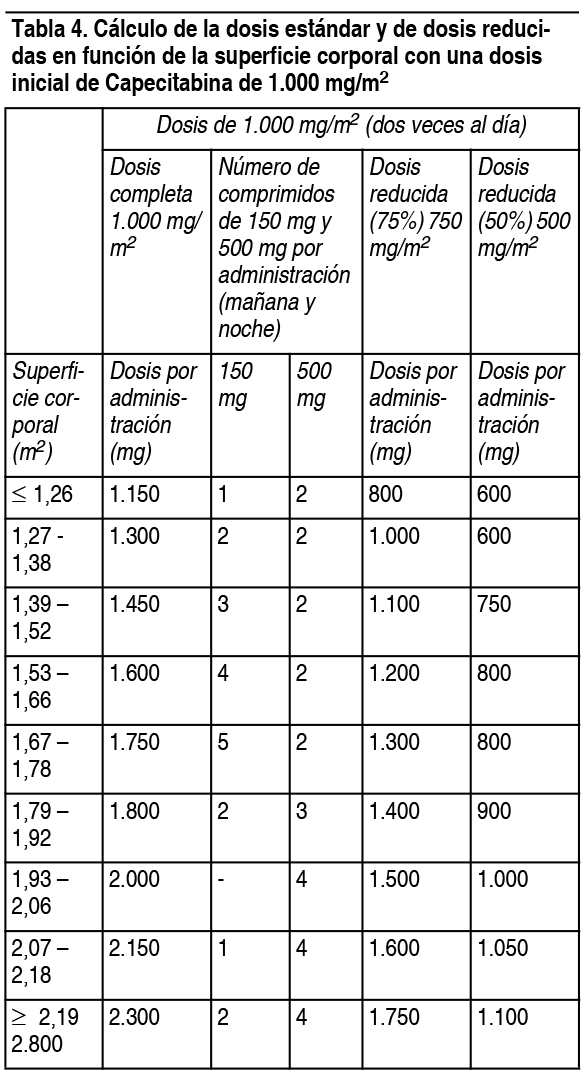
Posología:Dosis habitual: Los comprimidos de Xenevia 500 deben ingerirse enteros, con agua, dentro de los 30 minutos siguientes a una comida. Monoterapia: Carcinoma de colon, colorrectal y de mama: La dosis inicial recomendada de Xenevia 500 en monoterapia es de 1.250 mg/m2 2 veces al día (mańana y noche; equivalente a una dosis diaria total de 2.500 mg/m2) durante 2 semanas, seguido de 7 días de reposo farmacológico. Politerapia: Carcinoma de mama: En asociación con docetaxel, la dosis recomendada de Xenevia 500 es de 1.250 mg/m2 2 veces al día durante 2 semanas, seguido de 7 días de reposo farmacológico, y la del docetaxel, de 75 mg/m2 en infusión intravenosa (I.V.) de 1 hora, cada 3 semanas. De acuerdo con la información sobre el docetaxel, en los pacientes tratados con Xenevia 500 junto con docetaxel debe iniciarse una premedicación antes de administrarse el docetaxel. Carcinoma de colon, colorrectal, gástrico y esofagogástrico: En politerapia (excepto con irinotecán), la dosis inicial recomendada de Xenevia 500 es de 800-1.000 mg/m2 2 veces al día durante 2 semanas, seguido de 7 días de reposo farmacológico, o 625 mg/m2 2 veces al día si la administración es continua. En lo que respecta a la combinación con irinotecán, la dosis inicial recomendada de Xenevia 500 es de 800 mg/m2 administrados 2 veces al día durante 2 semanas, seguido de un período de descanso de 7 días en combinación con irinotecán en una dosis de 200 mg/m2 el día 1 de cada ciclo de 3 semanas. La inclusión de bevacizumab en un régimen politerápico no afecta la dosis inicial de Xenevia 500. Para pacientes con carcinoma de colon en estadio III se recomienda el tratamiento adyuvante durante un total de 6 meses. De acuerdo con la información sobre el cisplatino y el oxaliplatino, en los pacientes tratados con Xenevia 500 + cisplatino u oxaliplatino debe iniciarse una premedicación antiemética y suficientemente hidratante antes de administrarse el cisplatino. La dosis de capecitabina se calcula en función de la superficie corporal. En las tablas siguientes se muestran ejemplos de cálculos de la dosis estándar y de dosis reducidas con una dosis inicial de capecitabina de 1.250 mg/m2 o 1.000 mg/m2.
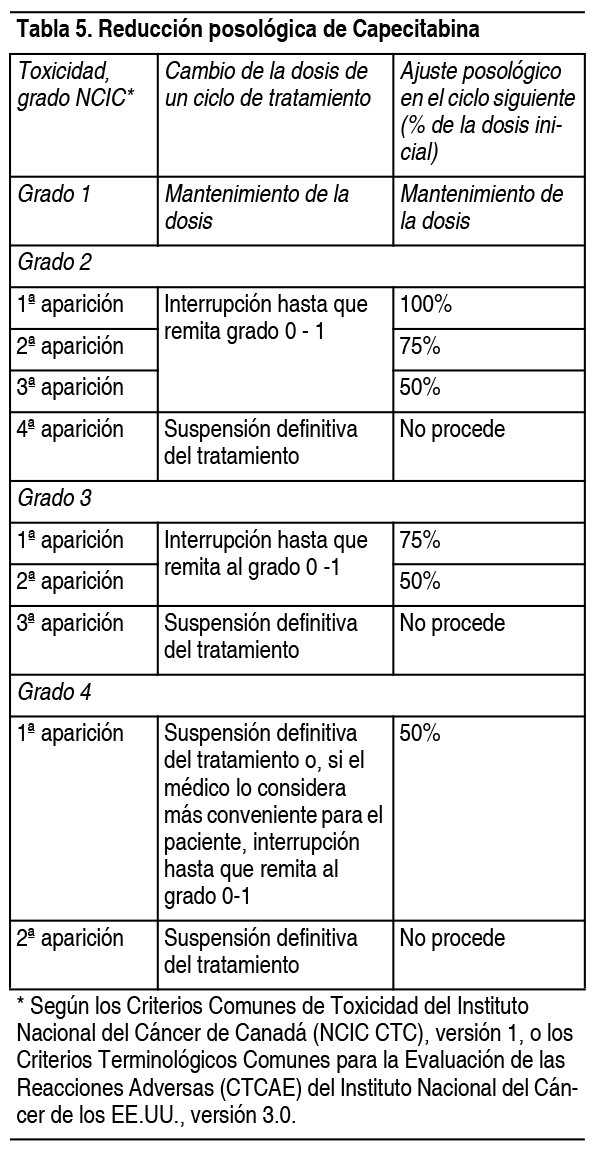
Ajustes posológicos durante el tratamiento: Información general: La toxicidad de capecitabina se puede controlar mediante tratamiento sintomático y/o modificando la dosis de capecitabina (interrupción de la medicación o reducción posológica). Una vez reducida la dosis, no deberá incrementarse después en ningún momento. Si el médico considera improbable que un efecto tóxico pueda volverse grave o poner en peligro la vida del paciente, podrá continuar el tratamiento con la misma dosis sin reducción o interrupción. En presencia de acontecimientos adversos de grado 1 no se recomienda ajustar la dosis. Si los acontecimientos adversos son de grado 2 ó 3, debe interrumpirse la administración de capecitabina. Una vez que el acontecimiento adverso haya desaparecido o disminuido de intensidad al grado 1, puede restablecerse el tratamiento con la dosis completa de capecitabina o, si es preciso, ajustada según la tabla 5. De presentarse una reacción de grado 4, se retirará capecitabina hasta su resolución o disminución al grado 1, para reinstaurar después su administración con la mitad de la dosis original. A los pacientes en tratamiento con capecitabina se les debe informar sobre la necesidad de interrumpir inmediatamente la administración si experimentan toxicidad moderada o grave. No se sustituirán las dosis de capecitabina omitidas por toxicidad. Hematología: No se administrará capecitabina a pacientes con un recuento basal de neutrófilos <1.5 x 109/l y/o de plaquetas <100 x 109/l. Si un análisis de laboratorio no programado durante un ciclo de tratamiento revela toxicidad hematológica de grado 3 ó 4, se retirará la capecitabina. La tabla siguiente recoge las modificaciones posológicas recomendadas en caso de toxicidad asociada a capecitabina.
Politerapia general: Los cambios posológicos por toxicidad cuando capecitabina se utilice en asociación con otros tratamientos deben realizarse según la tabla 3 en lo que respecta a capecitabina y de acuerdo con la información para el prescriptor del otro u otros compuestos. Si al comienzo de un ciclo de tratamiento debe posponerse la administración de capecitabina o del otro u otros compuestos, se pospondrá la administración de todos los medicamentos hasta que se den los requisitos para comenzar con todos. Si el médico considera que la toxicidad durante un ciclo de tratamiento no está relacionada con capecitabina, debe mantenerse la administración de capecitabina y ajustarse la dosis del otro u otros medicamentos de acuerdo con la respectiva información para el prescriptor. Si es necesario retirar definitivamente el otro u otros compuestos, se podrá reanudar el tratamiento con capecitabina cuando se den las condiciones para ello. Esta recomendación es válida para todas las indicaciones y todas las poblaciones especiales. Pautas posológicas especiales: Pacientes con insuficiencia hepática debida a metástasis hepáticas: En caso de insuficiencia hepática leve o moderada a causa de metástasis hepáticas, no es necesario ajustar la dosis inicial. Ahora bien, a tales pacientes se los debe vigilar estrechamente (ver Farmacocinética en poblaciones especiales y Advertencias y precauciones). En pacientes con insuficiencia hepática grave no se han realizado estudios. Insuficiencia renal: En los pacientes con una insuficiencia renal basal moderada (aclaramiento de creatinina de 30-50 ml/min [Cockroft y Gault] se recomienda disminuir la dosis de capecitabina al 75% de la dosis inicial de 1.250 mg/m2. En los pacientes con un ligero deterioro de la función renal (aclaramiento de creatinina de 51-80 ml/min) no se recomienda reducir la dosis inicial. Si se presenta algún acontecimiento adverso de grado 2, 3 ó 4, se recomienda interrumpir inmediatamente el tratamiento y mantener en estrecha vigilancia al paciente; después, se ajustará la dosis como se indica en la tabla 3 (ver Farmacocinética en poblaciones especiales). Si el aclaramiento calculado de creatinina desciende durante el tratamiento por debajo de 30 ml/min, se retirará la capecitabina. Las recomendaciones sobre el ajuste de la dosis en pacientes con deterioro moderado de la función renal son válidas tanto para la monoterapia como para el uso politerápico. Para el cálculo de la dosis, ver tablas 1 y 2. Nińos: No se han estudiado la seguridad y la eficacia de capecitabina en los nińos. Ancianos: No se necesita a justar la dosis inicial de capecitabina en monoterapia. Ahora bien, los efectos adversos de grado 3 ó 4 relacionados con el tratamiento han sido más frecuentes en pacientes de más de 80 ańos que en los más jóvenes. Cuando se ha utilizado capecitabina en asociación con otros compuestos, los pacientes ancianos (≥65 ańos) han experimentado más reacciones adversas (RA) de grado 3 ó 4 y más RA que han obligado a suspender la administración que los pacientes más jóvenes. Por tanto, se aconseja una cuidadosa vigilancia de los pacientes ancianos. En asociación con docetaxel: se ha observado una mayor incidencia de acontecimientos adversos de grado 3 ó 4 relacionados con el tratamiento y de acontecimientos adversos graves relacionados con el tratamiento entre los pacientes de 60 o más ańos. Para los pacientes de 60 o más ańos tratados con la asociación de capecitabina y docetaxel se aconseja empezar el tratamiento con una dosis de capecitabina reducida al 75% (950 mg/m2 2 veces al día). Para el cálculo de la dosis, ver tabla 1. Capecitabina en asociación con irinotecán: en los pacientes de 65 o más ańos se recomienda reducir la dosis inicial de capecitabina a 800 mg/m2 2 veces al día.
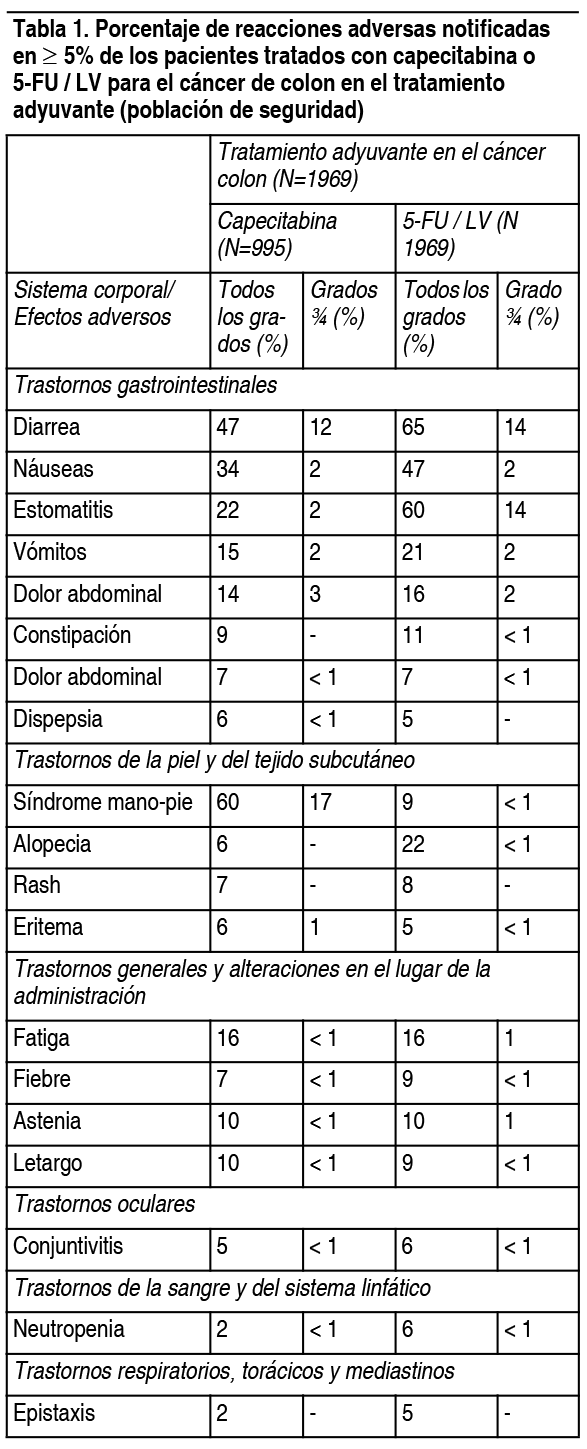
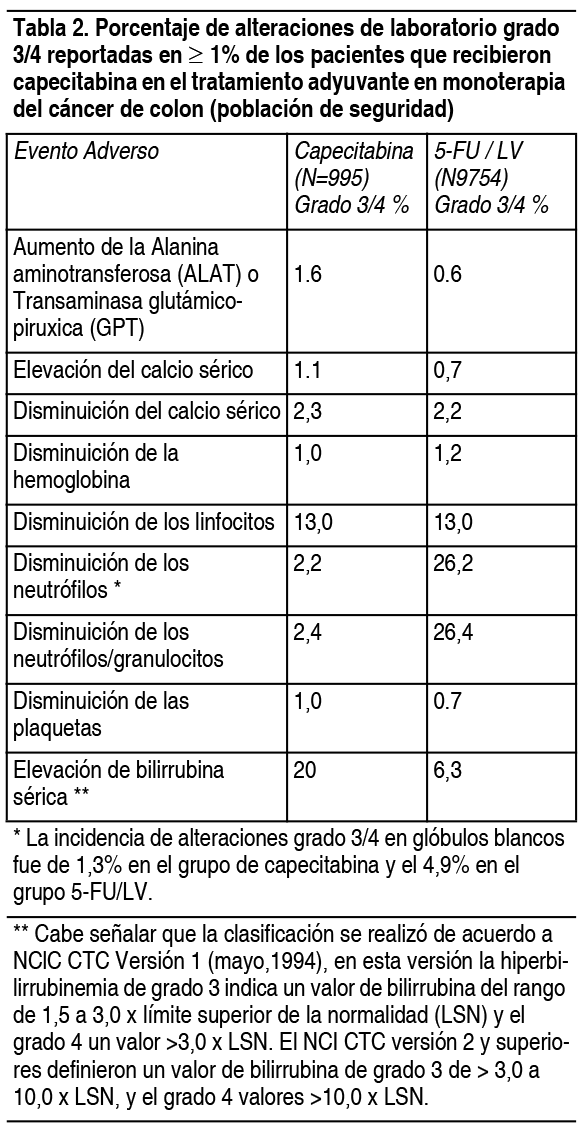
Efectos Colaterales: Debido a que los estudios clínicos se llevan a cabo en condiciones muy variables, las tasas de reacciones adversas medicamentosas (RAMs) observadas no pueden compararse directamente con las tasas de los ensayos clínicos de otro fármaco y puede que no reflejen las tasas observadas en la práctica. Capecitabina como adyuvante en el cáncer de colon: En la tabla 1 se incluyen las RAMs asociadas con el uso de capecitabina en monoterapia como adyuvante en el cáncer de colon (Dukes C), que ocurrieron en ≥5% de los pacientes de un estudio clínico de fase 3, que recibieron al menos 1 dosis de capecitabina y tenían por lo menos 1 evaluación de seguridad. Un grupo de pacientes fueron tratados con capecitabina 1250 mg/m2 2 veces al día administrados durante 2 semanas seguido de 1 semana de descanso, al resto de los pacientes se les administró 5-FU y leucovorin los días 1 y 5 cada 28 días. La duración media del tratamiento fue de 164 días, un 11% interrumpieron el tratamiento debido a reacciones adversas. Un 0.8% fallecieron por todas las causas, ya sea estando en el estudio o dentro de los 28 días posteriores de haber dejado de recibir el medicamento. La Tabla 2 muestra alteraciones de laboratorio grado 3/4 que ocurren en ≥1% de los pacientes de un ensayo fase 3 en pacientes con cáncer de colon (Dukes C) que recibieron al menos 1 dosis de capecitabina y tenían por lo menos 1 evaluación de seguridad.
Capecitabina como monoterapia en el cáncer colorrectal metastásico: Las reacciones adversas que ocurren en ≥5% de los pacientes en estudios en los que se administró capecitabina en primera línea para tratar cáncer colorrectal metastásico. Un grupo de pacientes fueron tratados con capecitabina y al resto de los pacientes se les administró 5-FU y leucovorina, la duración media del tratamiento fue de 139 días para los pacientes tratados con capecitabina y 140 días para los pacientes tratados con 5-FU/LV. Un 13% y 11% de los pacientes discontinuaron el tratamiento debido a reacciones adversas o enfermedades intercurrentes, respectivamente. Se produjeron un total de 82 muertes por todas las causas, ya sea mientras se realizaba el estudio o dentro de los 28 días de recibir el medicamento del estudio, siendo un 8.4% de los pacientes asignados al azar a capecitabina y 5.4% del grupo tratados con 5-FU/LV. Capecitabina en terapia de combinación con docetaxel en el cáncer de mama: Cuando se realizó el tratamiento conjunto con capecitabina y docetaxel, la duración media del tratamiento fue de 129 días en el grupo de combinación y 98 días en el grupo de monoterapia. Un 26% en el grupo de combinación y 19% en el grupo de monoterapia discontinuaron el tratamiento debido a reacciones adversas. El porcentaje de pacientes que requirieron reducciones de dosis debidas a reacciones adversas fue del 65% en el grupo de combinación y un 36% en el grupo de monoterapia. Las interrupciones del tratamiento fueron parte del plan de modificación de la dosis para el grupo de tratamiento combinado, pero no para los tratados con docetaxel en monoterapia.
Contraindicaciones: Hipersensibilidad a capecitabina o a 5-fluorouracilo. En pacientes con probada deficiencia de dihidropirimidina dehidrogenasa (DPD). En pacientes con insuficiencia renal grave (aclaramiento de creatinina por debajo de 30 ml/min). Durante el embarazo y la lactancia. En pacientes con leucopenia, neutropenia o trombocitopenia graves. En pacientes con insuficiencia hepática grave. En pacientes con insuficiencia renal grave (aclaramiento de creatinina por debajo de 30 ml/min). Tratamiento con sorivudina o sus análogos químicamente relacionados, tal como la brivudina.
Precauciones:Precauciones y advertencias especiales de empleo: General: Xenevia 500 solamente debe ser prescrito y monitorizado su tratamiento por un clínico con experiencia en el empleo de medicamentos antineoplásicos. La mayoría de los efectos adversos son reversibles y no necesariamente requieren de la discontinuación del tratamiento, aunque en ciertos casos puede ser necesaria un ajuste de dosis o suspensión (ver Posología). Los efectos tóxicos que limitan la dosis incluyen diarrea, dolor abdominal, náuseas, estomatitis y el síndrome mano-pie (reacción cutánea mano-pie, eritrodisestesia palmo-plantar). Diarrea: Capecitabina puede producir diarrea, en algunos casos severa. Se debe monitorizar cuidadosamente a los pacientes con diarrea severa y administrarles fluidos y reposición de electrolitos si llegaran a deshidratarse. Se pueden emplear los tratamientos antidiarreicos estándar (ej. loperamida). La NCIC CTC define la diarrea de grado 2 como un aumento de 4 a 6 deposiciones/día o deposiciones nocturnas, diarrea de grado 3 como un aumento de 7 a 9 deposiciones/día o incontinencia y malabsorción, y diarrea de grado 4 como un aumento de ≥10 deposiciones/día o melenas o la necesidad de soporte parenteral. La reducción de dosis se realizará según sea necesario (ver Posología). Si se observa una diarrea de grado 2 (o mayor, hasta 4), deberá interrumpirse de inmediato el tratamiento con capecitabina y se corregirá la deshidratación. No se reiniciará el tratamiento hasta que no se haya rehidratado al paciente y se hayan corregido y controlado las causas desencadenantes o hasta que se retroceda a la intensidad grado 1 de diarrea. Se han reportado casos de enterocolitis necrotizante. Coagulopatías: En los pacientes que reciban tratamiento concomitante con capecitabina y anticoagulantes derivados de cumarina por vía oral se debe monitorizar estrechamente su respuesta anticoagulante (INR o tiempo de protrombina) y se ajustará convenientemente la dosis de anticoagulante (ver Interacciones). Cardiotoxicidad: Se ha asociado la cardiotoxicidad con la terapia con fluoropirimidinas y capecitabina, la cual incluye infarto de miocardio, angina, disrrítmias, insuficiencia cardíaca, shock cardiogénico, muerte súbita, cardiomiopatía y cambios en el electrocardiograma (incluidos los casos muy raros de la prolongación QT). Estas reacciones adversas fueron más comunes en pacientes con un historial previo de enfermedad arterial coronaria. Deficiencia de dihidropirimidina dehidrogenasa (DPD): Raramente, se ha asociado con el 5-fluoroacilo (5-FU) toxicidad grave e inesperada por una deficiencia en la actividad del DPD, (por ejemplo, estomatitis, diarrea, neutropenia y neurotoxicidad). No se puede excluir ya que existe una relación entre los niveles bajos de DPD y el aumento de efectos tóxicos y potencialmente graves del 5-FU. Los pacientes con probada deficiencia de DPD no deben ser tratados con capecitabina. Insuficiencia renal: En pacientes con insuficiencia renal moderada se recomienda administrar una dosis reducida (ver Posología). En pacientes con insuficiencia renal moderada se recomienda monitorizar cuidadosamente al paciente para evaluar efectos adversos que pudieran ocurrir. Posteriores ajustes de dosis se recomiendan tal como está indicado en las tablas 2 y 3 (dependiendo del régimen) si un paciente desarrolla efectos adversos grado 2 a 4 (ver Advertencias y precauciones). Embarazo: La capecitabina puede causar dańo fetal si se administra a mujeres embarazadas. La administración de capecitabina produjo mortalidad embrionaria y teratogenia en los estudios sobre toxicidad reproductora en los animales. Estos datos constituyen efectos previsibles de los derivados de la fluoropirimidina. Capecitabina está contraindicado durante el embarazo. Si este fármaco es administrado durante el embarazo o la paciente quedara embarazada durante el tratamiento con capecitabina, deberla advertirse al paciente del potencial riesgo de efectos nocivos sobre el feto. Síndrome mano-pie: También conocido como reacción toxica cutánea mano-pie, eritrodisestesia palmo-plantar o eritema acral inducido por quimioterapia. El tiempo medio de aparición de este fenómeno fue de 79 días (rango de 11 a 360 días), con un rango de severidad de los grados 1 a 3 para los pacientes que recibieron monoterapia con capecitabina en el tratamiento del cáncer metastásico. El síndrome mano-pie de grado 1 está caracterizado por los siguientes síntomas: entumecimiento, disestesia/parestesia, hormigueo, tumefacción indolora o eritema de las manos y/o los pies y/o incomodidad que no altera las actividades normales del paciente. El síndrome mano-ple de grado 2 se define como un eritema doloroso y tumefacción de manos y/o pies produciendo una incomodidad que afecta a las actividades de la vida diaria del paciente. El síndrome mano-pie de grado 3 se define como una descamación húmeda, ulceración, aparición de vesículas y dolor intenso de manos y/o pies y/o fuerte malestar que causa incapacidad para trabajar o realizar las actividades de la vida diaria. Si se presenta un síndrome mano-pie de grado 2 ó 3, se debe interrumpir la administración de capecitabina hasta que desaparezca el efecto o disminuya en intensidad a grado 1. Después del síndrome mano-pie de grado 3, las dosis posteriores de capecitabina deben disminuirse (ver Posología). Hiperbilirrubinemia: Se han reportado casos en los que ha aumentado el registro de bilirrubina en sangre, este generalmente ha estado asociado a la existencia previa de metástasis hepáticas. Se debe interrumpir la administración de capecitabina en caso de que se presenten aumentos relacionados con el tratamiento de la bilirrubina >3.0 x ULN o de las aminotransferasas hepáticas (ALT, AST) de >2.5 x ULN. El tratamiento con capecitabina en monoterapia se podrá reanudar si la bilirrubina desciende hasta ≤3.0 x ULN o las aminotransferasas hepáticas disminuyen hasta ≤2.5 x ULN (ver Posología). Hematología: Los pacientes con un recuento basal de neutrófilos < 1.5 x 109/l y/o recuento de trombocitos < 100 x 109/l no deberán ser tratados con capecitabina. Si alguna prueba de laboratorio no prevista es realizada durante un ciclo de tratamiento y se observa que el recuento de neutrófilos cae por debajo de 1.0 x 109/l o que el recuento de plaquetas cae por debajo de 75 x 109/l, se debe interrumpir el tratamiento con capecitabina. Pacientes de edad avanzada: Las reacciones adversas de grado 3 ó 4 relacionadas con el tratamiento fueron más frecuentes en pacientes ≥60 ańos si se compara con pacientes más jóvenes. Se aconseja una cuidadosa monitorización de los pacientes ≥60 ańos. Hipo- o hipercalcemia: Se ha observado hipo- o hipercalcemia durante el tratamiento con capecitabina. Se debe tener precaución en pacientes con hipo- o hipercalcemia preexistente. Enfermedad del sistema nervioso central o periférico: Se debe tener precaución en pacientes con enfermedad del sistema nervioso central o periférico. p.ej. metástasis cerebrales o neuropatía. Diabetes mellitus o alteraciones de los electrolitos: Se debe tener precaución en pacientes con diabetes mellitus o con alteración de los electrolitos ya que estos pueden agravarse durante el tratamiento con capecitabina. Insuficiencia hepática: Cuando se administre capecitabina en pacientes con insuficiencia hepática de leve a moderada debido a metástasis hepáticas deberían ser cuidadosamente monitorizados. No se dispone de información relativa a su efecto en pacientes con insuficiencia hepática severa. Uso en poblaciones especiales: Embarazo y lactancia. Embarazo: Categoría D. No existen estudios adecuados sobre capecitabina en mujeres embarazadas, sin embargo, cabe seńalar que capecitabina puede causar dańo fetal si se administra a mujeres embarazadas. La administración de capecitabina produjo mortalidad embrionaria y teratogenia en los estudios sobre toxicidad reproductora en los animales. Estos datos constituyen efectos previsibles de los derivados de la fluoropirimidina. Capecitabina está contraindicada durante el embarazo: Si este fármaco es utilizado durante la gestación o si la paciente quedara embarazada durante el tratamiento con capecitabina, se deberá informar a la paciente acerca del potencial riesgo sobre el feto. Se debe aconsejar a las mujeres en edad fértil para evitar el embarazo mientras son tratadas con capecitabina. Durante el tratamiento se debe utilizar un tratamiento eficaz de anticoncepción. Lactancia: Se desconoce si capecitabina se elimina en la leche materna. En ratones lactantes se han detectado cantidades considerables de capecitabina y sus metabolitos en la leche. Se debe interrumpir la lactancia mientras se reciba tratamiento con capecitabina. Población pediátrica: No existe una recomendación de uso específica para capecitabina en la población pediátrica para las indicaciones de cáncer de colon colorrectal, gástrico y de mama, no se han establecido su seguridad y eficacia en menores de 18 ańos. Pacientes de edad avanzada: Los médicos tratantes deberían llevar una cuidadosa monitorización de los efectos adversos de capecitabina en pacientes ≥60 ańos. Insuficiencia hepática: Cuando se administre capecitabina en pacientes con insuficiencia hepática de leve a moderada debido a metástasis hepáticas deberían ser cuidadosamente monitorizados. No se dispone de información relativa a su efecto en pacientes con insuficiencia hepática severa. Insuficiencia renal: En pacientes con insuficiencia renal moderada (aclaramiento de creatinina basal de 30-50 ml/min) y severo aclaramiento de creatinina basal de <30 ml/min), se ha observado una mayor exposición de capecitabina, 5-FU, por lo que se recomienda administrar una menor dosis (ver Contraindicaciones; ver Posología; ver Advertencias y precauciones; ver Farmacología clínica). En pacientes con insuficiencia renal moderada se recomienda monitorizar cuidadosamente al paciente para evaluar efectos adversos que pudieran ocurrir. Efectos sobre la capacidad para conducir y utilizar máquinas: La influencia de capecitabina sobre la capacidad para conducir y utilizar máquinas es pequeńa o moderada. Capecitabina puede causar mareos, fatiga y náuseas.
Interacciones Medicamentosas:Interacciones con otros medicamentos y otras formas de interacción: Anticoagulantes derivados de cumarina: Se han reportado alteraciones de los parámetros de coagulación y/o sangrado en pacientes tratados con capecitabina concomitantemente con anticoagulantes derivados de cumarina tales como warfarina o fenprocumon. Estas reacciones se producen en algunos días hasta varios meses tras iniciar la terapia con capecitabina y, en unos pocos casos, dentro del primer mes tras finalizar el tratamiento con capecitabina. Estos eventos ocurrieron en pacientes con o sin metástasis hepáticas. Se observó que al administrar una dosis única de 20 mg de warfarina, el tratamiento con capecitabina puede aumentar el AUC de la warfarina un 57% y el valor de INR, un 91%. Estos resultados parecen indicar que la capecitabina y sus metabolitos inhiben la isoenzima 2C9 del citocromo P450, pero no tiene efecto sobre las isoenzimas 1A2 y 3A4. Aquellos pacientes que tomen anticoagulantes derivados de cumarina concomitantemente con capecitabina deben monitorizarse de forma regular para detectar alteraciones en sus parámetros de coagulación (TP o INR) y la dosis del anticoagulante se ajustara convenientemente. Fenitoína: Se debe monitorizar regularmente a aquellas pacientes que tomen fenitoína concomitantemente con capecitabina para detectar cualquier aumento en la concentración plasmática de fenitoína, puede ser necesario la reducción de la dosis de fenitoína. Se ha observado un incremento en las concentraciones plasmáticas de fenitoína que, en casos aislados, ha conllevado síntomas de intoxicación durante el uso concomitante de capecitabina con fenitoína. Se presume que la causa de esta elevación en las concentraciones plasmáticas de fenitoína sería debida a que capecitabina y sus metabolitos inhiben la isoenzima 2C9 del citocromo P450. Leucovorina: Las concentraciones plasmáticas de 5-FU y su toxicidad son incrementadas por la leucovorina. Se han informado muertes por enterocolitis severa, diarrea y deshidratación en pacientes ancianos que recibieron semanalmente leucovorina y 5-FU. Sustratos del citocromo P450 2C9: Aparte de la warfarina, no hay descritas interacciones entre la capecitabina y otros sustratos del CYP2C9. Se debe tener especial cuidado cuando se coadministra capecitabina y sustratos de CYP 2C9 (p. ejemplo la fenitoína). Sorivudina y análogos: se ha descrito una interacción medicamento-medicamento clínicamente significativa entre la sorivudina y el 5-FU originada por la inhibición de la dihidropirimidin dehidrogenasa por la sorivudina. Esta interacción, que provoca un aumento de la toxicidad de la fluoropirimidina, es potencialmente fatal. Por lo tanto, capecitabina no debe administrarse junto con sorivudina o sus análogos químicamente relacionados, tal como la brivudina. Debe existir al menos un período de espera de 4 semanas entre el fin del tratamiento con sorivudina o sus análogos químicamente relacionados, tal como la brivudina y el comienzo de la terapia con capecitabina. Interacción con alimentos: La administración con alimentos disminuye el índice de absorción de capecitabina (ver Farmacología clínica). En todos los ensayos clínicos se les dio instrucción a los pacientes para tomar capecitabina dentro de los 30 minutos después de una comida. Como los datos actuales de seguridad y eficacia están basados en la administración con alimentos, se recomienda administrar capecitabina con alimentos (ver Posología).
Sobredosificación: En caso de sobredosis o ingestión accidental, consulte inmediatamente a su médico. Las manifestaciones de sobredosis agudas incluyen náuseas, vómitos, diarrea, mucositis, irritación gastrointestinal y sangrado, así como depresión de la médula ósea. El manejo médico de la sobredosis debe incluir terapia individualizada e intervención médica de soporte encaminadas a corregir las manifestaciones clínicas y prevenir sus posibles complicaciones. La diálisis podría ser de utilidad en la reducción de las concentraciones de los metabolitos circulantes (5'-dFUR). Una dosis simple de capecitabina superior a 2000 mg/kg no fue mortal para ratones, ratas y monos (2.4; 4.8 y 9.6 veces la dosis recomendada en el hombre en base a la superficie corporal).
Conservación: Mantener a temperatura inferior a 30° C.
 Laboratorio
Laboratorio