Composición: Cada cápsula contiene: Fenofibrato 160 mg; Pravastatina Sódica 40 mg. Excipientes: Carbonato Ácido de Sodio, Lactosa Monohidrato, Ascorbil Palmitato, Celulosa Microcristalina, Povidona, Almidón Glicolato de Sodio (Tipo A), Estearato de Magnesio, Talco, Triacetina, Lauril Macrogolglicéridos, Macrogol 20.000, Hiprolosa, Gelatina, Óxido de Hierro Negro, Óxido de Hierro Amarillo, Dióxido de Titanio, Colorante Azul FD&C N° 2.
Indicaciones: Fibrotina Lidose está indicado como complemento para dietas y otros tratamientos no farmacológicos (por ejemplo, ejercicio, reducción de peso) para el tratamiento de la hiperlipidemia mixta en pacientes adultos con un alto riesgo cardiovascular para reducir los triglicéridos y aumentar el nivel del colesterol HDL cuando los valores de colesterol LDL se controlan suficientemente cuando reciben tratamiento con pravastatina 40 mg en monoterapia.
Propiedades:Farmacología: Mecanismo de acción: Fibrotina Lidose contiene fenofibrato y pravastatina, que tienen mecanismos de acción diferentes y efectos aditivos en la reducción de los lípidos séricos. Fenofibrato: El fenofibrato es un derivado del ácido fíbrico cuyos efectos hipolipemiantes descritos en el ser humano están mediados por la activación de los receptores activados por proliferadores de peroxisomas tipo alfa (PPARα). Los estudios de los efectos del fenofibrato en las fracciones de lipoproteínas indican un descenso de los valores de colesterol LDL y colesterol VLDL. Los valores de colesterol HDL se elevan con frecuencia. Los valores de LDL, VLDL y triglicéridos disminuyen. El efecto global es una menor proporción entre las lipoproteínas de baja y muy baja densidad y las lipoproteínas de alta densidad. Las propiedades hipolipemiantes del fenofibrato observadas en la práctica clínica se han explicado in vivo en ratones transgénicos y en cultivos de hepatocitos humanos por la activación de los receptores activados por proliferadores de peroxisomas tipo alfa (PPARα). Por medio de este mecanismo, el fenofibrato aumenta la lipólisis y elimina del plasma las partículas ricas en triglicéridos debido a la activación de la lipoproteína lipasa y a la inhibición de la producción de la apoproteína C-III. La activación de los receptores PPARα aumenta también la síntesis de apoproteínas A-I, A-II y colesterol HDL. La concentración plasmática de ácido úrico aumenta aproximadamente un 20% en los pacientes con hiperlipidemia, especialmente en los que presentan la enfermedad tipo IV. El fenofibrato tiene un efecto uricosúrico y, por consiguiente, ofrece un beneficio adicional a esos pacientes. Pravastatina: La pravastatina es un inhibidor competitivo de la 3-hidroxi-3-metilglutaril-coenzima A (HMG-CoA) reductasa, la enzima que cataliza el paso inicial limitante de la biosíntesis del colesterol, y produce un efecto hipolipemiante por 2 vías. En primer lugar, reduce ligeramente la síntesis del colesterol intracelular como consecuencia de la inhibición reversible y competitiva específica de la HMG-CoA reductasa. El resultado es un incremento del número de receptores LDL en la superficie celular y un aumento del catabolismo mediado por esos receptores y del aclaramiento del colesterol LDL circulante. En segundo lugar, la pravastatina inhibe la producción de LDL al inhibir la síntesis hepática de colesterol VLDL, precursor del colesterol LDL. Tanto en sujetos sanos como en pacientes con hipercolesterolemia, la pravastatina reduce los valores de colesterol total, colesterol LDL, apolipoproteína B, colesterol VLDL y triglicéridos; y eleva los valores de colesterol HDL y apolipoproteína A. Fibrotina Lidose: Los efectos respectivos de la pravastatina y el fenofibrato son complementarios. La pravastatina es más eficaz para conseguir un descenso de los valores de colesterol LDL y colesterol total, pero sus efectos en los TG y el colesterol HDL son modestos, mientras que el fenofibrato es muy eficaz para conseguir un descenso de los TG y una elevación del colesterol HDL, pero tiene pocos efectos en el colesterol LDL. Por su parte, los fibratos tienen la propiedad de que modifican el tamańo y la densidad de las partículas de colesterol LDL para hacerlo menos aterógeno. Se ha demostrado también que la combinación de fibratos y estatinas aumenta de manera sinérgica las actividades de transcripción de los receptores PPARα. Perfil farmacocinético: No se observaron interacciones farmacocinéticas clínicamente significativas cuando se administró fenofibrato conjuntamente con pravastatina. Absorción: En un estudio de dosis únicas, se demostró la bioequivalencia de Fibrotina Lidose y la administración conjunta de fenofibrato y pravastatina. Sin embargo, los resultados de un estudio de dosis múltiples indicaron que el producto no es bioequivalente porque la biodisponibilidad tras la administración reiterada es un 20% menor para el componente fenofibrato de la combinación. Esa diferencia se debe al contenido de grasas de la comida. Por consiguiente, no se puede considerar que la CDF (Fibrotina) sea intercambiable por la administración conjunta de sendos preparados de fenofibrato y pravastatina como únicos principios activos. Se ha realizado un estudio farmacocinético después de la administración de una dosis única de Fibrotina Lidose en condiciones postprandiales y de ayuno. Los resultados de dicho estudio indican que los alimentos afectan tanto a la velocidad como al grado de absorción en la CDF. La biodisponibilidad de ácido fenofíbrico es menor en condiciones de ayuno después de la administración de una dosis única de la combinación de fenofibrato-pravastatina 160/40 mg. La disminución de los valores de AUCt, AUC∞ y Cmáx del ácido fenofíbrico (estimación puntual) es del 30.94%, el 10.9% y el 68.71% respectivamente. La biodisponibilidad de la pravastatina es mayor tras la administración de una dosis única del producto de ensayo fenofibrato/pravastatina 160/40 mg en condiciones de ayuno que tras la administración de una dosis única de dicho producto en condiciones postprandiales. El aumento de AUC∞, AUCt y Cmáx es del 111.88 %, 114.06 % y 115.28 %, respectivamente. Al igual que con otras formulaciones que contienen fenofibrato, se recomienda tomar la combinación fija con alimentos, ya que la biodisponibilidad del fenofibrato aumenta cuando se administra con alimentos, sin que se altere la eficacia hipolipemiante de la pravastatina. Pravastatina: La pravastatina se administra por vía oral en su forma activa. Se absorbe rápidamente y las concentraciones plasmáticas máximas se alcanzan entre 1 y 1.5 horas después de su ingestión. Por término medio, se absorbe el 34% de la dosis administrada por vía oral, con una biodisponibilidad absoluta del 17%. La presencia de alimentos en el tubo digestivo reduce la biodisponibilidad, pero el efecto hipocolesterolemiante de la pravastatina es el mismo se tome con o sin alimentos. Tras su absorción, el 66% de la pravastatina experimenta un metabolismo de primer paso en el hígado, que es su principal lugar de acción y el lugar principal de la síntesis de colesterol y del aclaramiento de colesterol LDL. Los estudios in vitro han demostrado que la pravastatina es transportada al interior de los hepatocitos en mucha mayor medida que al interior de otras células. Debido a este importante metabolismo de primer paso en el hígado, las concentraciones plasmáticas de pravastatina tienen sólo un valor limitado en la predicción del efecto hipolipemiante. Las concentraciones plasmáticas son proporcionales a las dosis administradas. Fenofibrato: Las concentraciones plasmáticas máximas (Cmáx) se alcanzan en el plazo de 4 a 5 horas después de la administración oral. Las concentraciones plasmáticas se mantienen estables si el paciente recibe un tratamiento continuo. La absorción de fenofibrato aumenta cuando se administra conjuntamente con alimentos. El efecto de los alimentos aumenta con el contenido de grasas: cuanto mayor sea el contenido de lípidos, mayor es la biodisponibilidad del fenofibrato. Distribución: Pravastatina: Aproximadamente el 50% de la pravastatina circulante se une a las proteínas plasmáticas. El volumen de distribución es de aproximadamente 0.5 l/kg. Una pequeńa cantidad de pravastatina pasa a la leche materna. Fenofibrato: El ácido fenofíbrico se une fuertemente a la albúmina plasmática (más de 99%). Metabolismo y excreción: Pravastatina: La pravastatina no es metabolizada significativamente por el citocromo P450, ni parece ser un sustrato ni un inhibidor de la glicoproteína-P, pero sí es un sustrato de otras proteínas transportadoras. Tras su administración oral, el 20% de la dosis inicial se elimina en la orina y el 70% en las heces. La semivida de eliminación plasmática de la pravastatina oral es de 1.5 a 2 horas. Tras su administración intravenosa, el 47% de la dosis se elimina por vía renal y otro 53% por excreción biliar y biotransformación. El principal producto de degradación de la pravastatina es el metabolito isomérico 3-α-hidroxi, que exhibe entre la décima y la cuadragésima parte de la actividad inhibidora de la HMG CoA-reductasa que la del compuesto original. El aclaramiento sistémico de pravastatina es de 0.81 l/h/kg y el aclaramiento renal de 0.38 l/h/kg, lo que indica secreción tubular. Fenofibrato: No se puede detectar fenofibrato intacto en el plasma, donde el metabolito principal es el ácido fenofíbrico. El fármaco se elimina principalmente en la orina. Prácticamente todo el medicamento se elimina en 6 días. El fenofibrato se excreta principalmente en forma de ácido fenofíbrico y su conjugado glucurónido. En pacientes de edad avanzada, no se modifica el aclaramiento plasmático total aparente de ácido fenofíbrico. La semivida de eliminación plasmática del ácido fenofíbrico es de 20 horas aproximadamente. Los estudios cinéticos realizados tras la administración de una dosis única o un tratamiento continuo han demostrado que el fármaco no se acumula. El ácido fenofíbrico no se elimina mediante hemodiálisis.
Posología:Vía de administración: Vía oral. Dosis: Antes de iniciar el tratamiento con Fibrotina Lidose, se deben descartar otras causas secundarias de dislipidemia y prescribir a los pacientes una dieta estándar para reducir el colesterol y los triglicéridos, que deberán seguir durante todo el tratamiento. Dosis usual: Adultos: La dosis recomendada es de 1 cápsula al día. Los pacientes deben mantener las restricciones dietéticas instituidas antes del tratamiento. La respuesta al tratamiento debe vigilarse mediante la determinación de los valores de lípidos séricos. El tratamiento con Fibrotina Lidose suele ir seguido de una rápida reducción de dichos valores y, si en el plazo de 3 meses no ha conseguido una respuesta suficiente, deberá suspenderse. Adultos mayores: El tratamiento con Fibrotina Lidose se debe prescribir después de haber evaluado la función renal. Se dispone de datos limitados sobre la seguridad de Fibrotina Lidose en pacientes mayores de 75 ańos, por lo que se recomienda administrarlo con precaución. Pacientes pediátricos: El uso de Fibrotina Lidose en la población pediátrica no es relevante para la indicación de dislipidemia mixta. Pacientes con insuficiencia renal: No es necesario modificar la posología en pacientes con insuficiencia renal leve. Fibrotina Lidose está contraindicado en pacientes con insuficiencia renal moderada o grave (definida como un aclaramiento de creatinina <60 ml/min). Pacientes con insuficiencia hepática: No es necesario ajustar la dosis en pacientes con insuficiencia hepática leve. Fibrotina Lidose no está recomendado en pacientes con insuficiencia hepática moderada y está contraindicado en pacientes con insuficiencia hepática grave. Forma de administración: La dosis recomendada es de 1 cápsula diaria en la cena. Dado que con el estómago vacío se absorbe peor, Fibrotina Lidose debe tomarse con alimentos.
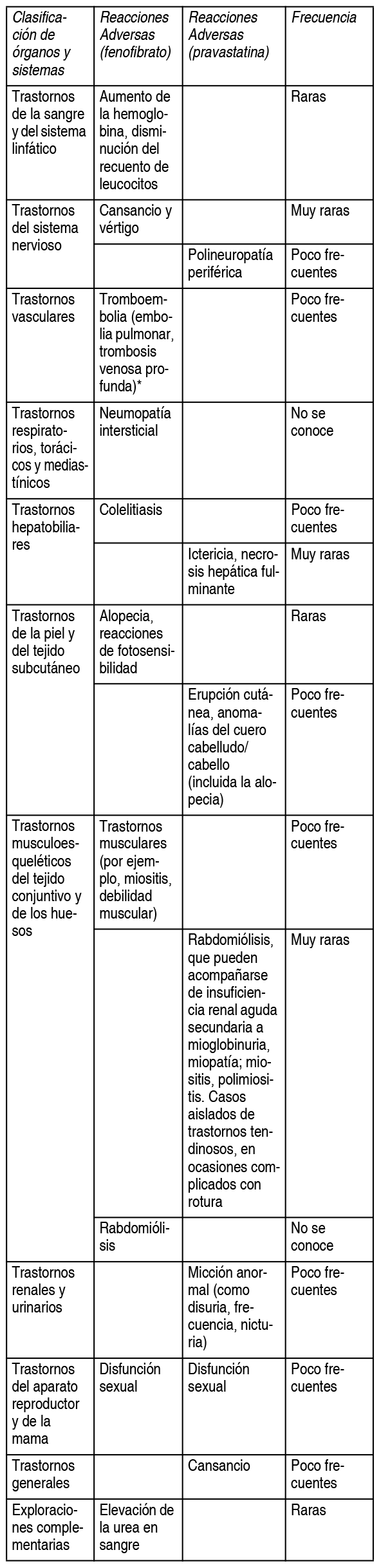
Efectos Colaterales:Se han observado y notificado las siguientes reacciones adversas con las siguientes frecuencias: muy frecuentes (≥1/10); frecuentes (≥1/100 a <1/10); poco frecuentes (≥1/1.000 a ≤1/100); raras (≥1/10.000 a ≤1/1.000); muy raras (≤1/10.000).Trastornos del sistema inmunológico: Poco frecuentes: Reacciones de hipersensibilidad. Trastornos del metabolismo y de la nutrición: Poco frecuentes: Diabetes mellitus agravada, obesidad. Trastornos psiquiátricos: Poco frecuentes: Alteración del sueńo como insomnio y pesadillas. Trastornos del sistema nervioso: Poco frecuentes: Mareo, cefalea, parestesia. Trastornos cardíacos: Poco frecuentes: Palpitaciones. Trastornos gastrointestinales: Frecuentes: Distensión abdominal, dolor abdominal, dolor abdominal superior, estreńimiento, diarrea, sequedad de boca, dispepsia, eructos, flatulencia, náuseas, molestias abdominales, vómitos. Trastornos hepatobiliares: Frecuentes: Elevación de transaminasas. Poco frecuente: Dolor hepático, elevación de gammaglutamiltransferasa. Trastornos de la piel y del tejido subcutáneo: Poco frecuentes: Prurito, urticaria. Trastornos musculoesqueléticos, del tejido conjuntivo y de los huesos: Poco frecuentes: Artralgia, dolor de espalda, elevación de creatina fosfocinasa sérica, espasmos musculares, dolor musculoesquelético, mialgia, dolor en las extremidades. Trastornos renales y urinarios: Poco frecuentes: Elevación de la creatinina sérica, disminución del aclaramiento renal de creatinina, aumento de aclaramiento renal de creatinina. Insuficiencia renal. Trastornos generales y alteraciones en el lugar de administración: Poco frecuentes: Astenia, cansancio, enfermedad pseudogripal. Exploraciones complementarias: Poco frecuentes:Elevación del colesterol en sangre, elevación de los triglicéridos en sangre, aumento de las lipoproteínas de baja densidad, ganancia de peso. Descripción de algunos acontecimientos adversos: Musculoesqueléticos: Rara vez se han notificado elevaciones marcadas y persistentes de la creatina fosfocinasa (CK). En estudios clínicos, la incidencia de elevaciones importantes de la creatina fosfocinasa (CK ≥3 veces el LSN, ≤5 veces el LSN) fue del 1.92% en los pacientes tratados con Fibrotina Lidose. En el 0.38% de los pacientes tratados con Fibrotina Lidose se observaron elevaciones clínicamente relevantes de la creatina fosfocinasa (CK ≥5 veces el LSN, ≤10 veces el LSN sin síntomas musculares). En el 0.06% de los pacientes tratados con Fibrotina Lidose se observaron elevaciones clínicamente relevantes (CK ≥10 veces el LSN sin síntomas musculares). Reacciones hepáticas: Rara vez se han notificado elevaciones marcadas y persistentes de las transaminasas séricas. En estudios clínicos, la incidencia de elevaciones de las transaminasas séricas (ALT o AST ≥3 veces el LSN, ≤5 veces el LSN) fue del 0.83% en los pacientes tratados con Fibrotina Lidose. En el 0.38% de los pacientes tratados con Fibrotina Lidose se observaron elevaciones clínicamente relevantes de las transaminasas séricas (ALT o AST ≥5 veces el LSN). Información adicional sobre los principios activos respectivos de la combinación en dosis fijas: Fibrotina Lidose contiene pravastatina y fenofibrato. A continuación se indican otras reacciones adversas relacionadas con otros medicamentos que contienen pravastatina o fenofibrato y observadas en ensayos clínicos y en la experiencia posterior a la comercialización, y que podrían ocurrir con Fibrotina Lidose. Las categorías de frecuencia se basan en la información de la Ficha Técnica o Resumen de las características del Producto de la pravastatina y el fenofibrato disponibles.
*En el estudio FIELD (estudio del fenofibrato), un ensayo aleatorizado y controlado con placebo realizado en 9795 pacientes con diabetes mellitus tipo 2, se observó un aumento estadísticamente significativo de los casos de pancreatitis en los pacientes que recibieron fenofibrato frente a los que recibieron placebo (0.8% frente al 0.5%; p=0.031). En ese mismo estudio, se notificó un incremento estadísticamente significativo de la incidencia de trombosis venosa profunda (1.0% [48/4900 sujetos] en el grupo de placebo frente a 1.4 en el grupo de fenofibrato [67/4895 pacientes]; p = 0.074). Con algunas estatinas se han comunicado los siguientes acontecimientos adversos: Pesadillas, pérdida de memoria, depresión, casos excepcionales de enfermedad pulmonar intersticial, especialmente con tratamientos de larga duración. Diabetes mellitus: La frecuencia dependerá de la presencia o ausencia de factores de riesgo (glucemia en ayunas ≥5.6 mmol/l, IMC >30 kg/m2, hipertrigliceridemia, antecedentes de hipertensión arterial.
Contraindicaciones:Fibrotina Lidose está contraindicado en los siguientes casos: Hipersensibilidad a los principios activos o a cualquiera de los componentes de este medicamento. Insuficiencia hepática grave como cirrosis biliar o hepatopatía activa, con elevaciones persistentes e inexplicadas de los resultados obtenidos en las pruebas de la función hepática (incluida la elevación de las transaminasas séricas) más de 3 veces por encima del límite superior de la normalidad (LSN). Insuficiencia renal moderada o grave (definida como un aclaramiento estimado de creatinina < 60 ml/min.) Reacción fotoalérgica o fototóxica conocida durante el tratamiento con fibratos o ketoprofeno. Enfermedad de la vesícula biliar. Pancreatitis aguda o crónica, con excepción de la pancreatitis aguda secundaria a hipertrigliceridemia intensa. Antecedentes personales de miopatía o rabdomiólisis con estatinas o fibratos, o elevación confirmada de la creatina fosfocinasa (CK) más de 5 veces por encima del límite superior de la normalidad (LSN) con un tratamiento previo de estatinas. Embarazo y lactancia. Nińos y adolescentes (menores de 18 ańos).
Precauciones: Las propiedades farmacocinéticas de Fibrotina Lidose no son totalmente idénticas a las observadas con la administración conjunta de las monoterapias existentes junto con una comida rica en grasas o en condiciones de ayuno. Los pacientes no deben pasar de la administración conjunta de sendos preparados de fenofibrato y pravastatina a Fibrotina Lidose. Trastornos musculoesqueléticos y del tejido conjuntivo: Al igual que con otros fármacos hipolipemiantes, la pravastatina y el fenofibrato se han asociado a la aparición de mialgia, miopatía y, muy raramente, rabdomiólisis con o sin insuficiencia renal secundaria. La rabdomiólisis es un trastorno agudo y potencialmente mortal de la musculatura esquelética, que puede aparecer en cualquier momento durante el tratamiento y se caracteriza por una destrucción muscular masiva asociada a una elevación importante de la CK (normalmente entre 30 y 40 veces por encima del LSN) que termina produciendo mioglobinuria. El riesgo de toxicidad muscular aumenta con la administración conjunta de un fibrato y un inhibidor de la 3-hidroxi-3-metil-glutaril-coenzima A (HMG-CoA) reductasa. En todos los pacientes que presenten síntomas musculares inexplicados, como dolor o hipersensibilidad, debilidad muscular o calambres musculares, se tiene que considerar la posibilidad de una miopatía y se recomienda medir los valores de CK. Por consiguiente, antes de iniciar el tratamiento se debe sopesar con cuidado la relación entre el beneficio potencial y el riesgo de Fibrotina Lidose y vigilar en los pacientes la aparición de signos de toxicidad muscular. Algunos factores de predisposición, como una edad > 70 ańos, insuficiencia renal, insuficiencia hepática, hipotiroidismo, antecedentes personales de toxicidad muscular con una estatina o un fibrato, antecedentes personales o familiares de enfermedades musculares hereditarias o abuso de alcohol, pueden aumentar el riesgo de toxicidad muscular, por lo que en esos pacientes se recomienda medir los valores de CK antes de iniciar el tratamiento combinado. No se debe administrar simultáneamente pravastatina con ácido fusídico sistémico. Se han notificado casos de rabdomiólisis (con algunos casos de fallecimientos) en pacientes que recibieron esta combinación. Se debe interrumpir el tratamiento con estatinas a los pacientes para los que el uso sistémico de ácido fusídico sea imprescindible, durante todo el período de tratamiento con el ácido fusídico. Se debe advertir al paciente de que acuda al médico de inmediato si nota algún síntoma de debilidad, dolor o sensibilidad muscular. El tratamiento con estatinas se podrá reiniciar transcurridos 7 días tras la última dosis de ácido fusídico. En casos excepcionales en los que se precise un tratamiento sistémico prolongado con ácido fusídico (p. ej., para el tratamiento de infecciones graves), únicamente se debe considerar la administración simultánea de pravastatina y ácido fusídico caso por caso y bajo una supervisión médica intensiva. Antes del inicio del tratamiento: Se deben medir los valores de CK antes del inicio del tratamiento. Los valores basales de CK obtenidos antes del inicio del tratamiento pueden servir de referencia en el caso de que se produzca posteriormente una elevación durante el tratamiento combinado. Los valores obtenidos de CK deben interpretarse teniendo en cuenta otros factores potenciales que pueden causar dańo muscular transitorio, como ejercicio físico intenso o traumatismo muscular, y la medición de estos valores debe repetirse cuando sea necesario. Si el valor basal de CK está significativamente elevado más de 5 veces por encima del LSN, se tendrá que repetir la medición al cabo de 5-7 días. Si se confirma la elevación, el tratamiento no se podrá instaurar definitivamente (ver sección Contraindicaciones). Durante el tratamiento: En todos los pacientes se recomienda realizar controles de rutina de la CK cada 3 meses durante los primeros 12 meses de tratamiento combinado y posteriormente con la frecuencia que el médico considere oportuna. Se debe pedir a los pacientes que informen a su médico de inmediato si presentan dolor, hipersensibilidad, debilidad o calambres musculares inexplicados. En esos casos deben medirse los valores de CK. Si se detecta y confirma una marcada elevación de los valores de CK (más de 5 veces el LSN), tendrá que interrumpirse el tratamiento con Fibrotina Lidose. Se debe considerar también la interrupción del tratamiento si los síntomas musculares son intensos y causan un malestar continuo (con independencia de cuáles sean los valores de CK). Si se sospecha una enfermedad muscular hereditaria en esos pacientes, no se recomienda reanudar el tratamiento con Fibrotina Lidose. Se han notificado, en muy raras ocasiones, casos de miopatía necrotizante inmunomediada (MNIM) durante o después del tratamiento con algunas estatinas. Clínicamente, la MNIM se caracteriza por debilidad muscular proximal persistente y elevación de la creatina kinasa sérica, que persisten a pesar de la suspensión del tratamiento con la estatina. Trastornos hepatobiliares: Al igual que con otros productos hipolipemiantes, se han observado incrementos moderados de los valores de las transaminasas en algunos pacientes tratados con pravastatina o fenofibrato. En la mayoría de los casos, las transaminasas vuelven a su valor basal sin necesidad de suspender el tratamiento. Se recomienda vigilar los valores de las transaminasas cada 3 meses durante los 12 primeros meses de tratamiento y posteriormente con la frecuencia que el médico considere necesaria. Los pacientes con elevación de las transaminasas deben recibir una atención especial y suspender el tratamiento si la elevación de la aspartato aminotransferasa (AST) y la alanina aminotransferasa (ALT) excede en más de 3 veces el LSN y es persistente. Se recomienda precaución cuando se administre Fibrotina Lidose a pacientes con antecedentes de hepatopatía o consumo elevado de alcohol. Pancreatitis: Se han notificado algunos casos de pancreatitis en pacientes tratados con fenofibrato o pravastatina. Ese hecho puede deberse a su falta de eficacia en pacientes con hipertrigliceridemia intensa, a un efecto directo del medicamento o a un fenómeno secundario mediado por la formación de cálculos o sedimentos en los conductos biliares que producen la obstrucción del colédoco. Trastornos renales y urinarios: Fibrotina Lidose está contraindicado en la insuficiencia renal moderada o grave. En todos los pacientes se recomienda evaluar el aclaramiento de creatinina al inicio del tratamiento, cada 3 meses durante los 12 primeros meses de tratamiento combinado y posteriormente con la frecuencia que el médico considere necesaria. El tratamiento debe suspenderse si se estima que el aclaramiento de la creatinina es mayor de 60 ml/min. Neumopatía intersticial: Se han notificado casos excepcionales de neumopatía intersticial con algunas estatinas, especialmente en tratamientos de larga duración. Las manifestaciones iniciales pueden consistir en disnea, tos no productiva y deterioro del estado de salud general (fatiga, pérdida de peso y fiebre). Si se sospecha que un paciente presenta neumopatía intersticial, se debe suspender el tratamiento con Fibrotina Lidose. Colelitiasis: El fenofibrato puede aumentar la eliminación de colesterol en la bilis y llegar a producir una colelitiasis. Si se sospecha la presencia de colelitiasis, habrá que realizar estudios de la vesícula biliar. La administración de Fibrotina Lidose debe interrumpirse si se encuentran cálculos biliares. Episodios venotromboembólicos: En el estudio FIELD se observó un incremento estadísticamente significativo de la incidencia de embolia pulmonar (0.7% en el grupo de placebo frente a 1.1% en el grupo de fenofibrato; p = 0.022) y un incremento estadísticamente no significativo de la incidencia de trombosis venosa profunda (1.0% en el grupo de placebo (48/4900 pacientes) frente a 1.4% en el grupo de fenofibrato (67/4895); p = 0.074). Ese mayor riesgo de episodios trombóticos venosos puede deberse a un aumento de la concentración de homocisteína, que es un factor de riesgo para la trombosis y otros factores no identificados. La importancia clínica de este hecho no está clara. Por consiguiente, se recomienda precaución en pacientes con antecedentes de embolia pulmonar. Diabetes mellitus: Algunas evidencias sugieren que las estatinas elevan la glucemia y en algunos pacientes, con elevado riesgo de desarrollar diabetes, pueden originar niveles de hiperglucemia que requieran cuidados propios de la diabetes. Este riesgo, sin embargo es en parte contrarrestado por la reducción del riesgo vascular que se obtiene con las estatinas y, por tanto, no debe haber razón para suprimir el tratamiento. Los pacientes de riesgo (glucemia en ayunas 5.6 a 6.9 mmol/l, IMC > 30 kg/m2, hipertrigliceridemia e hipertensión) deberán ser monitorizados tanto clínica como bioquímicamente de acuerdo con las guías clínicas apropiadas. Efectos sobre la capacidad para conducir y utilizar máquinas: La influencia de Fibrotina Lidose sobre la capacidad para conducir y utilizar máquinas es nula o insignificante. No obstante, si es necesario conducir vehículos o utilizar máquinas, hay que tener en cuenta que se pueden sufrir mareos y alteraciones visuales durante el tratamiento. Embarazo y lactancia: Embarazo: Pravastatina sódica: La pravastatina está contraindicada durante el embarazo y debe administrarse a mujeres en edad fértil sólo cuando estas pacientes tengan escasas probabilidades de concebir y hayan sido informadas de los posibles riesgos. Se recomienda especial precaución en mujeres en edad fértil y siempre debe comprobarse que hayan entendido correctamente los posibles riesgos que conlleva el tratamiento con pravastatina durante el embarazo. Si una paciente tiene previsto quedar o se queda embarazada, debe informar al médico de inmediato y suspender el tratamiento con pravastatina debido al posible riesgo para el feto. Fenofibrato: No hay datos sobre el uso del fenofibrato en pacientes embarazadas. Los estudios realizados en animales no han demostrado efectos teratógenos. Se ha demostrado toxicidad fetal con dosis que están dentro del intervalo de la toxicidad materna. Se desconoce el riesgo potencial para el ser humano. Fibrotina Lidose: No se dispone de datos sobre el uso combinado de pravastatina y fenofibrato en mujeres embarazadas. Esta combinación no se ha evaluado en estudios de toxicidad para la reproducción. Se desconoce el riesgo potencial para el ser humano. Por consiguiente, teniendo en cuenta que la pravastatina está contraindicada durante el embarazo Fibrotina Lidose también lo está. Lactancia: Pravastatina sódica: Una pequeńa cantidad de pravastatina se excreta en la leche materna humana; por consiguiente, la pravastatina está contraindicada durante la lactancia. Fenofibrato: El fenofibrato se excreta en la leche de ratas hembra. No existen datos sobre la excreción del fenofibrato o de sus metabolitos en la leche materna humana. Fibrotina Lidose: No se han realizado estudios de Fibrotina Lidose en animales lactantes. Por consiguiente, teniendo en cuenta que la pravastatina está contraindicada durante la lactancia, Fibrotina Lidose también lo está. Fertilidad: No se han observado efectos en la fertilidad del fenofibrato o de la pravastatina en estudios de toxicidad para la reproducción. No se dispone de datos relativos a la fertilidad con el uso combinado de fenofibrato y pravastatina.
Interacciones Medicamentosas: No se han realizado estudios formales de interacciones con Fibrotina Lidose; sin embargo, el uso simultáneo de los principios activos en pacientes que estaban participando en estudios clínicos no ha producido interacciones inesperadas. A continuación se presenta la información disponible sobre los principios activos administrados en monoterapia (fenofibrato y pravastatina). Interacciones con pravastatina: Colestiramina/colestipol: La administración concomitante redujo en aproximadamente el 40% o el 50% la biodisponibilidad de la pravastatina. No se produjo ninguna disminución clínicamente significativa de la biodisponibilidad ni del efecto terapéutico cuando la pravastatina se administró 1 hora antes o 4 horas después que la colestiramina, o 1 hora antes que el colestipol. Ciclosporina: La administración concomitante de pravastatina y ciclosporina multiplica aproximadamente por cuatro la exposición sistémica a la pravastatina. En algunos pacientes, sin embargo, el aumento de dicha exposición puede ser mayor. Se recomienda la vigilancia clínica y bioquímica de los pacientes que reciban esa combinación. Productos metabolizados por el citocromo P450: La pravastatina no es metabolizada en un grado clínicamente significativo por el sistema del citocromo P450. Por ello se pueden ańadir medicamentos que son metabolizados por el sistema del citocromo P450 o que lo inhiben a un régimen estable de pravastatina sin alterar significativamente la concentración plasmática de esta última, como ocurre con otras estatinas. La ausencia de una interacción farmacocinética significativa con pravastatina se ha demostrado específicamente para una serie de medicamentos, sobre todo los que son sustratos o inhibidores de la CYP3A4, como diltiazem, verapamilo, itraconazol, ketoconazol, inhibidores de la proteasa o zumo de pomelo, y los inhibidores de la CYP2C9 (por ejemplo, fluconazol). En 1 de 2 estudios sobre interacciones con pravastatina y eritromicina, se observó un incremento estadísticamente significativo del área bajo la curva (AUC) (70%) y de la Cmáx (121%) en el grupo de pravastatina. En un estudio similar con claritromicina, se observó un incremento estadísticamente significativo del AUC (110%) y de la Cmáx (127%). Aunque esos cambios fueron pequeńos, se recomienda precaución cuando se administre pravastatina en combinación con eritromicina o claritromicina. Ácido fusídico: Una interacción medicamentosa entre la pravastatina y el ácido fusídico puede provocar un aumento del riesgo de rabdomiólisis. La administración conjunta de ácido fusídico y estatinas podría aumentar el riesgo de miopatías, inclusive la rabdomiólisis. La administración conjunta de esta combinación puede provocar un aumento de las concentraciones plasmáticas de los 2 fármacos. Todavía no se conoce el mecanismo de esta interacción (si se trata de un mecanismo farmacodinámico o farmacocinético, o ambos). Se han notificado casos de rabdomiólisis (con algunos casos de fallecimientos) en pacientes que recibieron esta combinación. En el caso de que el tratamiento con ácido fusídico sea necesario, se debe interrumpir el tratamiento con pravastatina mientras dure el tratamiento con el ácido fusídico. Otros medicamentos: En estudios de interacciones no se observaron diferencias estadísticamente significativas en la biodisponibilidad cuando la pravastatina se administró con ácido acetilsalicílico, antiácidos (administrados 1 hora antes que la pravastatina), ácido nicotínico o probucol. Interacciones con el fenofibrato: Resinas secuestradoras de ácidos biliares: Las resinas secuestradoras de ácidos biliares reducen con frecuencia la absorción de otros medicamentos y cuando se administran conjuntamente con fenofibrato, éste debe tomarse 1 hora antes, o entre 4 y 6 horas después, para que las resinas no interfieran con la absorción de fenofibrato. Anticoagulantes orales: El fenofibrato potencia el efecto anticoagulante oral y puede aumentar el riesgo de hemorragia. Se recomienda reducir la dosis de anticoagulantes en casi 1/3 al inicio del tratamiento, para luego ajustarla gradualmente en caso necesario de acuerdo con los controles del cociente internacional normalizado (INR). Por lo tanto, no se recomienda esta combinación. Ciclosporina: Se han notificado algunos casos graves de insuficiencia renal reversible durante la administración conjunta de fenofibrato y ciclosporina. Por consiguiente, en estos pacientes se recomienda vigilar estrechamente la función renal e interrumpir el tratamiento con fenofibrato si se produce una alteración significativa de los parámetros clínicos. Interacción con los alimentos: Fibrotina Lidose debe tomarse con las comidas, puesto que los alimentos aumentan la biodisponibilidad del fenofibrato. En todos los ensayos clínicos, los pacientes recibieron instrucciones de tomar Fibrotina Lidose diariamente con la cena y de mantener las restricciones dietéticas instituidas antes del tratamiento. Puesto que los datos disponibles sobre seguridad y eficacia se basan en la administración con alimentos y con restricciones dietéticas, se recomienda tomar Fibrotina Lidose con alimentos.
Sobredosificación: En caso de sobredosis, se debe administrar un tratamiento sintomático e instituir las medidas de apoyo adecuadas. Tratamiento general de la sobredosis: Pravastatina: Los casos declarados de sobredosis fueron asintomáticos y no alteraron los valores obtenidos en los análisis. No se conoce ningún antídoto específico. Fenofibrato: No se conoce ningún antídoto específico. El fenofibrato no se elimina mediante hemodiálisis.
 Laboratorio
Laboratorio