Composición: Cada cápsula contiene: 75 mg, 100 mg o 125 mg de Palbociclib Base Libre. Excipientes: Celulosa Microcristalina, Lactosa Monohidrato, Almidón Glicolato Sódico, Dióxido de Silicio Coloidal, Estearato de Magnesio y Cubiertas de Cápsula de Gelatina Dura. Las cubiertas de cápsulas opacas de color anaranjado claro, anaranjado claro/caramelo y caramelo contienen: Gelatina, Oxido de Hierro Rojo, Oxido de Hierro Amarillo y Dióxido de Titanio. La tinta de impresión contiene: Goma Shellac, Dióxido de Titanio, Alcohol Isopropílico, Hidróxido de Amonio, Alcohol N-butílico, Propilenglicol y Simeticona.
Indicaciones: Ibrance® está indicado en combinación con letrozol para tratar a mujeres post-menopáusicas con cáncer de mama avanzado con receptor de estrógeno (ER) positivo, de receptor 2 del factor de crecimiento epidérmico humano (HER2) negativo como tratamiento inicial endocrino para su enfermedad metastásica.
Posología: La dosis recomendada de Ibrance® es de 1 cápsula de 125 mg, administrada por vía oral 1 vez al día durante 21 días consecutivos, seguidos de 7 días sin tratamiento (cronograma 3/1) para completar un ciclo de 28 días en combinación con letrozol de 2.5 mg 1 vez al día, administrado continuamente durante el ciclo de 28 días. Ibrance® debe ser ingerido con alimentos. Es necesario instar a los pacientes a tomar su dosis aproximadamente a la misma hora todos los días. Continúe el tratamiento mientras el paciente obtenga como resultado un beneficio clínico a partir de la terapia. Si el paciente vomita u omite una dosis, no se debe administrar una dosis adicional ese día. La siguiente dosis prescrita se debe administrar a la hora habitual. Las cápsulas de Ibrance® deben ingerirse completas (no se deben masticar, aplastar ni abrir antes de su ingestión). No se debe ingerir ninguna cápsula si estuviera rota, rajada o si tuviera signos de no estar intacta. Se recomienda la modificación de la dosis de Ibrance® en función de la seguridad individual y la tolerabilidad. El tratamiento de algunas reacciones adversas puede requerir la interrupción o demora temporal de la dosis y/o reducciones de dosis o la interrupción permanente, según los cronogramas de reducción de dosis de las Tablas 1, 2 y 3 (consulte la sección advertencias y precauciones y la sección Efectos colaterales. Tabla 1: Modificaciones de las dosis recomendadas para Ibrance®en caso de eventos colaterales: Nivel de dosis: Dosis. Dosis recomendada: 125 mg/día. Primera reducción de dosis: 100 mg/día. Segunda reducción de dosis: 75 mg/día*. * Si se requiere otra reducción de dosis por debajo de 75 mg/día, interrumpa el tratamiento. Tabla 2: Manejo y modificación de la dosis de Ibrance®: Toxicidades hematológicas: Grado de CTCAE: Modificaciones de la dosis: Grado 1 ó 2: No se requiere ajuste de la dosis. Grado 3b: No se requiere ajuste de la dosis. Considere repetir el monitoreo de hemograma completo 1 semana después. No inicie el siguiente ciclo hasta la recuperación al Grado ≥2. ANC Grado 3: (<1000 a 500/mm3). + Fiebre ≥38.5 şC y/o infección Suspenda Ibrance® y no inicie el siguiente ciclo hasta la recuperación al Grado 2 (1000/mm3). Reanude en la dosis inferior siguiente. Grado 4b: Suspenda Ibrance® y no inicie el siguiente ciclo hasta la recuperación al Grado ≥2. Reanude en la dosis inferior siguiente. Calificación según Versión 4.0 de CTCAE. ANC = recuento absoluto de neutrófilos; CTCAE = Criterios Comunes de Terminología para Eventos Adversos. aMonitoree el hemograma antes de iniciar el tratamiento con Ibrance® y al comenzar cada uno de los ciclos, así como también en el día 14 de los primeros 2 ciclos y según la indicación clínica. bExcepto la linfocitopenia (a menos que se relacione con eventos clínicos; por ejemplo, infecciones oportunistas). Tabla 3: Manejo y modificación de la dosis de Ibrance®: Toxicidades no hematológicas: Grado de CTCAE: Modificaciones de la dosis. Grado 1 ó 2: No se requiere ajuste de la dosis. Toxicidad no hematológica de Grado 3 (si persiste a pesar del tratamiento médico): Suspender hasta que se resuelvan los síntomas y pasen a ser de: Grado 1: 2. Grado 2 (si no es considerado un riesgo de seguridad para el paciente). Reanude en la dosis inferior siguiente. Calificación según la Versión 4.0 de CTCAE. CTCAE = Criterios Comunes de Terminología para Eventos Adversos. No se requieren modificaciones de dosis en función de la edad, el sexo o el peso corporal del paciente. Poblaciones especiales: Población de ancianos: No se requieren ajustes de dosis en pacientes de ≥65 ańos. Población pediátrica: No se han establecido la seguridad ni la eficacia de Ibrance® en nińos o adolescentes de ≥18 ańos. Deterioro hepático: No se requieren ajustes de dosis para pacientes con deterioro hepático leve (bilirrubina total ≥1 × límite superior normal [LSN] y aspartato aminotransferasa [AST] >1 × LSN o bilirrubina total de >1.0-1.5 × LSN y cualquier AST). No se han realizado estudios de Ibrance® en pacientes con deterioro hepático severo ni moderado (bilirrubina total >1.5 × LSN y cualquier AST). Deterioro renal: No se requieren ajustes de la dosis en pacientes con deterioro renal leve a moderado (eliminación de creatinina [CrCl] ≥30 ml/min). No se han realizado estudios en Ibrance® en pacientes con deterioro renal severo (CrCl <30 ml/min) ni que requieran hemodiálisis.
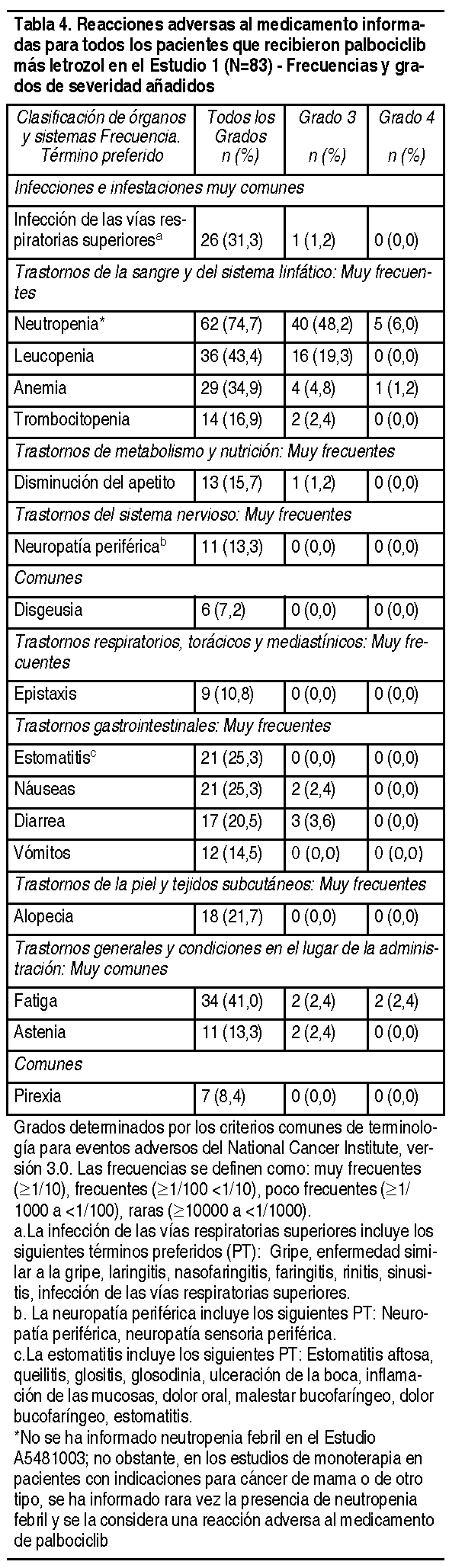
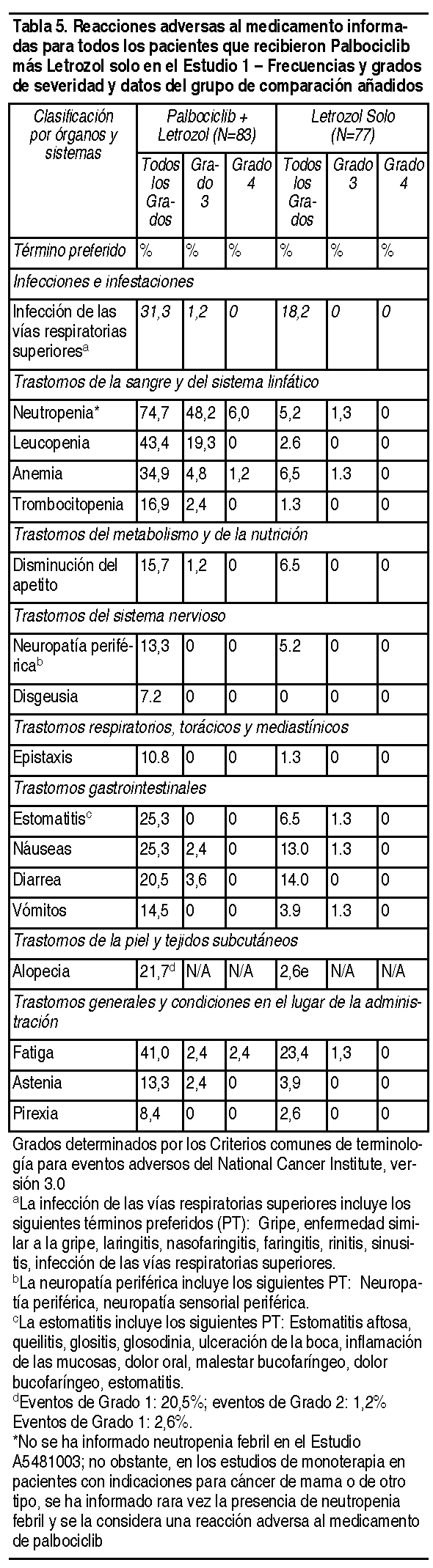
Efectos Colaterales: En el Estudio 1, se evaluó la seguridad de palbociclib. Un total de 165 pacientes con cáncer de mama avanzado con HER2 negativo y ER positivo fueron aleatorizadas 1:1 para recibir la combinación de palbociclib más letrozol o letrozol solo. Los datos que se describen a continuación reflejan la exposición de las 83 pacientes que recibieron al menos 1 dosis de la combinación y de las 77 pacientes que recibieron al menos 1 dosis de letrozol solo en la parte de la Fase 2 del Estudio 1. La duración mediana del tratamiento para palbociclib fue de 13.8 meses, mientras que la duración mediana del tratamiento para letrozol en el grupo de letrozol solo fue de 7.6 meses. Se produjeron reducciones de dosis a causa de una reacción adversa (según la lista de la Tabla 4) de cualquier grado en el 36% de los pacientes que recibieron palbociclib más letrozol. No se permitió una reducción de dosis para letrozol en el Estudio 1. La interrupción permanente debido a una reacción adversa ocurrió en 7 de 83 (8%) pacientes que recibieron palbociclib más letrozol y en 2 de 77 (2.6%) pacientes que recibieron letrozol solo. Las reacciones adversas al medicamento más comunes de cualquier grado informadas en los pacientes del grupo de palbociclib más letrozol fueron neutropenia, leucopenia, fatiga, anemia, infección de las vías respiratorias superiores*, náuseas, estomatitis*, alopecia, diarrea, trombocitopenia, disminución del apetito, vómitos, astenia, neuropatía periférica* y epistaxis (*términos agrupados definidos en la Tabla 4). La reacción adversa grave al medicamento más frecuentemente informada en los pacientes que recibían palbociclib más letrozol fue diarrea (2.4%). En general, se informó neutropenia de cualquier grado en 62 (74.7%) pacientes en el grupo de combinación; se informó neutropenia de Grado 3 en 40 (48.2%) pacientes y se informó neutropenia de Grado 4 en 5 (6.0%) pacientes (consulte la sección Advertencias y precauciones). Las reacciones adversas al medicamento informadas en pacientes que recibieron palbociclib más letrozol en el Estudio 1 se detallan en la Tabla 4. Las reacciones adversas (RAM) se detallan por clasificación de órganos y sistemas, y grados de severidad en las tablas de frecuencia de RAM y de categorías a continuación:
Contraindicaciones: Ninguna.
Advertencias:Neutropenia: Se ha observado una disminución del recuento de neutrófilos en los ensayos clínicos con Ibrance®. Se informaron disminuciones en los recuentos de neutrófilos de Grado 3 (56.6%) o 4 (4.8%) en pacientes que recibieron Ibrance® más letrozol en el ensayo clínico aleatorizado (Estudio 1). El tiempo mediano al primer episodio de neutropenia de cualquier grado según los datos de laboratorio fue de 15 días (13-117 días). La duración mediana de neutropenia de Grado ≥3 fue de 7 días. Se han informado eventos de neutropenia febril en el programa clínico de Ibrance®, aunque no se han observado casos de neutropenia febril en el Estudio 1. Monitoree el hemograma antes de iniciar el tratamiento con Ibrance® y al comenzar cada uno de los ciclos, así como en el Día 14 de los primeros 2 ciclos y según la indicación clínica. Se recomienda la interrupción, la reducción o el retraso de la dosis en los ciclos de tratamiento inicial en los pacientes que desarrollan neutropenia de Grado 3 ó 4 (consulte la Tabla 2). Infecciones: Dado que Ibrance® tiene propiedades mielosupresoras, es posible que predisponga a los pacientes a infecciones. Se han informado infecciones en una tasa más elevada de pacientes tratados con Ibrance® más letrozol, en comparación con pacientes tratados con letrozol solo, en el Estudio 1. Las infecciones de Grado 3 ó 4 se dieron en el 5% de los pacientes tratados con Ibrance® más letrozol, mientras que ningún paciente bajo letrozol solo presentó infección alguna de Grado 3 ó 4. Controle a los pacientes a fin de detectar signos y síntomas de infección, y trátelos según sea médicamente adecuado (consulte la sección Efectos colaterales). Los médicos deben indicar a los pacientes que deben informar de inmediato cualquier episodio de fiebre que se presente.
Precauciones:Fertilidad: En estudios no clínicos, no se presentaron efectos en el ciclo estral ni el apareamiento en ratas hembra. No obstante, no se han obtenido datos clínicos respecto de la fertilidad en mujeres. Según los hallazgos de seguridad no clínicos, es posible que la fertilidad en el hombre se encuentre comprometida por el tratamiento con Ibrance® (consulte la sección Toxicidad). Los hombres deberían considerar la preservación de su esperma antes de comenzar el tratamiento con Ibrance®. Mujeres en edad fértil/embarazadas: No existen estudios adecuados y bien controlados con Ibrance® en mujeres embarazadas. En función de los hallazgos realizados en animales y el mecanismo de acción, palbociclib puede dańar al feto si se lo administra a una mujer embarazada. En estudios en animales, palbociclib resultó teratogénico y fetotóxico en dosis tóxicas para la madre. Si se administra este fármaco durante el embarazo o si la paciente queda embarazada mientras recibe este medicamento, la paciente debe ser alertada del dańo potencial para el feto. A las mujeres en edad fértil se les debe indicar que eviten quedar embarazadas durante el tiempo que reciben palbociclib. Cuando tomen este medicamento, las mujeres en edad fértil o los hombres que reciben este medicamento y tienen parejas en edad fértil deben usar métodos anticonceptivos adecuados durante el tratamiento y durante 90 días como mínimo luego de haberlo finalizado. Lactancia: No se han realizado estudios en humanos para evaluar el efecto de Ibrance® sobre la producción de leche, su presencia en la leche materna o sus efectos en el nińo alimentado con leche materna. Se desconoce si palbociclib se excreta en la leche humana. Las pacientes que reciben palbociclib no deben amamantar. Efectos en la capacidad para conducir y utilizar maquinaria: No se han realizado estudios con Ibramce® sobre la capacidad de conducir vehículos y operar maquinaria. Sin embargo, los pacientes que presenten fatiga mientras toman Ibramce® deben tener precaución a la hora de conducir u operar maquinaria.
Interacciones Medicamentosas: Palbociclib se metaboliza principalmente mediante CYP3A y la enzima SULT2A1 de sulfotransferasa (SULT). In vivo, el palbociclib es un inhibidor temporal de CYP3A. Agentes que pueden aumentar las concentraciones plasmáticas de palbociclib: Efecto de los inhibidores de CYP3A: Los datos de un estudio de interacciones medicamentosas (DDI) en sujetos sanos indican que la coadministración de varias dosis de 200 mg de itraconazol con una sola dosis de 125 mg de Ibrance® aumentó la exposición total a palbociclib (área bajo la curva, AUCinf) y la exposición máxima (Cmáx) aproximadamente en un 87% y un 34%, respectivamente, en relación con una sola dosis de palbociclib de 125 mg administrado solo. Debe evitarse la administración concomitante de inhibidores potentes de CYP3A, entre ellos: amprenavir, atazanavir, boceprevir, claritromicina, conivaptan, delavirdina, diltiazem, eritromicina, fosamprenavir, indinavir, itraconazol, ketoconazol, lopinavir, mibefradil, miconazol, nefazodona, nelfinavir, posaconazol, ritonavir, saquinavir, telaprevir, telitromicina, verapamil, voriconazol, y toronja o jugo de toronja. Agentes que pueden disminuir las concentraciones plasmáticas de palbociclib: Efecto de los inductores de CYP3A: Los datos de un estudio de DDI en sujetos sanos indican que la coadministración de varias dosis diarias de rifampicina 600 mg con una dosis única de palbociclib 125 mg disminuyó el AUCinf y la Cmáx del palbociclib en un 85% y un 70%, respectivamente, en relación con una dosis única de palbociclib 125 mg administrado solo. Se debe evitar la administración concomitante de inductores potentes de CYP3A que incluyen, entre otros: carbamazepina, felbamato, nevirapina, fenobarbital, fenitoína, primidona, rifabutina, rifampicina, rifapentina y hierba de San Juan. Efecto de los agentes que reducen el ácido: Los datos de un estudio de DDI en sujetos sanos indicaron que la coadministración de una dosis única de palbociclib con varias dosis de rabeprazol PPI en sujetos alimentados disminuyó la Cmáx de palbociclib 125 mg en un 41%, pero tuvo un impacto limitado en el AUCinf (disminución del 13%) en comparación con una sola dosis de palbociclib administrado solo. Dado el efecto reducido en el pH gástrico de los antagonistas del receptor H2 y los antiácidos locales en comparación con los PPI, en pacientes alimentados, no se presentan efectos clínicos relevantes de PPI, antagonistas del receptor H2 ni antiácidos locales respecto de la exposición a palbociclib. Los datos de otro estudio de DDI en sujetos sanos indicaron que la coadministración de una sola dosis de palbociclib con varias dosis de rabeprazol PPI en sujetos en ayunas disminuyó el AUCinf y la Cmáx de palbociclib 125 mg en un 62% y un 80%, respectivamente, cuando se lo comparó con una sola dosis de palbociclib administrado solo. Por lo tanto, Ibrace® debería administrarse con alimentos (consulte la sección Posología). Efecto de Ibrance® en otros medicamentos: Palbociclib es un inhibidor temporal débil de CYP3A después de una administración de la dosis diaria de 125 mg en estado de equilibrio en seres humanos. En un estudio de interacciones medicamentosas en sujetos sanos, la coadministración de midazolam con varias dosis de palbociclib aumentó los valores del AUCinf y la Cmáx de midazolam en un 61% y un 37%, respectivamente, según se lo comparó con la administración de midazolam solo. In vitro, palbociclib no es un inhibidor de CYP1A2, 2A6, 2B6, 2C8, 2C9, 2C19 ni 2D6, ni es un inductor de CYP1A2, 2B6, 2C8 y 3A4 en concentraciones con relevancia clínica. Interacciones medicamentosas entre palbociclib y letrozol: Los datos de la parte de evaluación de interacciones medicamentosas de un estudio clínico en pacientes con cáncer de mama mostraron que no había interacciones medicamentosas entre palbociclib y letrozol cuando se coadministraban los 2 medicamentos. Efecto de tamoxifeno en la exposición a palbociclib: Los datos de un estudio de interacciones medicamentosas en sujetos masculinos sanos indicaron que las exposiciones del palbociclib eran comparables cuando se coadministraba una sola dosis de palbociclib con varias dosis de tamoxifeno y cuando se administraba exclusivamente palbociclib. Estudios in vitro con transportadores: Las evaluaciones in vitro indican que palbociclib tiene bajo potencial para inhibir las actividades de la glucoproteína P (P-gp) transportadoras del medicamento, la proteína de resistencia al cáncer de mama (BCRP), el transportador de aniones orgánicos (OAT)1, OAT3, el transportador de cationes orgánicos (OCT)2, el polipéptido transportador de aniones orgánicos (OATP)1B1, OATP1B3 y la bomba de exportación de sales biliares (BSEP) en concentraciones de relevancia clínica. Según los datos in vitro, es improbable que el transporte mediado por P- gp y BCRP influencie el alcance de la absorción oral de palbociclib en dosis terapéuticas.
Sobredosificación: No se conoce un antídoto para palbociclib. El tratamiento de la sobredosis de Ibrance® debe constar de medidas generales de apoyo. Toxicidad: Se identificó metabolismo de glucosa alterado (glucosuria, hiperglucemia, disminución de insulina) relacionado con cambios en el páncreas (vacuolización de células del islote), ojo (cataratas, degeneración de lentes), dientes (degeneración/necrosis de ameloblastos en dientes en crecimiento activo), rińón (vacuolización tubular) y tejido adiposo (atrofia) en el estudio toxicológico de repetición de dosis de 27 semanas en ratas, y estas afecciones prevalecieron más en machos en dosis ≥30 mg/kg/día (aproximadamente 9 veces la exposición humana [AUC] en la dosis recomendada). Algunos de estos hallazgos (glucosuria, hiperglucemia, vacuolización de células del islote y vacuolización tubular del rińón) estuvieron presentes en el estudio de toxicología de repetición de dosis de 15 semanas en ratas, aunque con incidencia y severidad más bajas. Las ratas que se utilizaron para este estudio tenían aproximadamente 7 semanas de vida al inicio de los estudios. No se identificaron metabolismo de glucosa alterado ni cambios relacionados en el páncreas, ojo, dientes, rińón y tejido adiposo en perros en estudios toxicológicos de repetición de dosis de hasta 39 semanas de duración. Carcinogenicidad: No se han llevado a cabo estudios de carcinogenicidad con palbociclib. Genotoxicidad: En un ensayo de mutación inversa bacteriana (Ames), no se observó una mutagénesis del palbociclib, y en el ensayo de aberración cromosómica de linfocitos humanos in vitro no indujo aberraciones cromosómicas estructurales. Palbociclib indujo presencia de micronúcleo mediante un mecanismo aneugénico en células de ovario de hámster chino in vitro y en la célula ósea de ratas macho en dosis ≥100 mg/kg/día. El nivel de efecto no observado para la aneugenicidad fue de aproximadamente 7 veces la exposición clínica humana según el AUC. Deterioro de la fertilidad: En un estudio de fertilidad y desarrollo embrionario temprano en ratas hembra, se administró palbociclib por vía oral durante 15 días antes de aparearse hasta el día 7 de embarazo, lo que no provocó toxicidad embrionaria en dosis de hasta 300 mg/kg/día con exposiciones maternales sistémicas de casi 4 veces la exposición humana (AUC) en la dosis recomendada. En estudios de desarrollo embriofetal en ratas y conejas, los animales preńados recibieron dosis orales de hasta 300 mg/kg/día y 20 mg/kg/día de palbociclib, respectivamente, durante el período de organogénesis. La dosis tóxica para la madre de 300 mg/kg/día fue fetotóxica en ratas, lo que provocó reducciones en los pesos corporales de los fetos. En dosis ≥100 mg/kg/día en ratas, hubo un incremento en la incidencia de una variación esquelética (mayor incidencia de una costilla presente en la séptima vértebra cervical). En la dosis tóxica para la madre de 20 mg/kg/día en conejas, hubo una mayor incidencia de variaciones esqueléticas, entre ellas falanges pequeńas en la extremidad anterior. En las dosis de 300 mg/kg/día en ratas y 20 mg/kg/día en conejas, las exposiciones maternales sistémicas fueron de aproximadamente 4 y 9 veces la exposición humana (AUC) en la dosis recomendada. No se han llevado a cabo estudios de fertilidad en machos; sin embargo, en los estudios de toxicidad con repetición de dosis, se observó un deterioro testicular en ratas y perros en dosis de 30 y 0.2 mg/kg/día, respectivamente (aproximadamente 8 y 0.1 veces la exposición clínica humana según la AUC, respectivamente). Se observó reversibilidad parcial de los efectos en los órganos reproductores masculinos en ratas y perros, luego de un período de no administración de la dosis de 4 y 12 semanas, respectivamente.
Observaciones: Ver información completa para prescribir en documento de monografía entregado por Pfizer Chile S.A.
 Laboratorio
Laboratorio