Composición: Comprimidos de color morado, biconvexos, ovales, marcados con "572 Tri" en un lado. Cada comprimido recubierto contiene: 50 mg de Dolutegravir como Dolutegravir Sódico, 600 mg de Abacavir como Sulfato de Abacavir y 300 mg de Lamivudina. Lista de excipientes: De acuerdo con fórmula aprobada en el registro sanitario.
Indicaciones: Triumeq está indicado para el tratamiento del virus de inmunodeficiencia humana tipo 1 (VIH -1). Limitaciones de uso: No se recomienda el uso de Triumeq en pacientes con historial de resistencia a cualquier componente de Triumeq. Triumeq en monoterapia, no se recomieda en pacientes con sustituciones de integrasa, asociadas a resistencia o sospecha clínica de resistencia a Inhibidor de la Transferencia de la Cadena de Integrasa (INSTI), debido a que la dosis de dolutegravir de Triumeq es insuficiente en estas poblaciones.
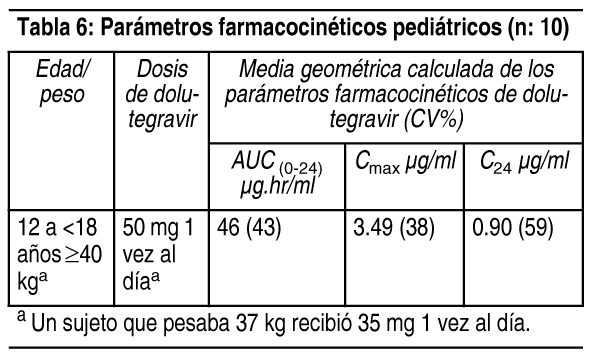
Propiedades:Farmacodinámica: Mecanismo de acción: Dolutegravir inhibe a la integrasa del VIH uniéndose al sitio activo de la integrasa y bloqueando el paso de transferencia de la hebra de la integración del ácido desoxirribonucleico (ADN) retroviral, el cual es esencial para el ciclo de replicación del VIH. Los ensayos bioquímicos de transferencia de la hebra utilizando la integrasa purificada del VIH 1 y un sustrato pre procesado de ADN, mostraron valores de IC50 de 2.7 nM y 12.6 nM. In vitro, dolutegravir se disocia lentamente del sitio activo del tipo salvaje del complejo integrasa- ADN (t ˝ 71 horas). Abacavir y lamivudina son INTR, y son inhibidores potentes y selectivos del VIH-1 y el VIH-2. Tanto abacavir como lamivudina son metabolizados de forma secuencial mediante cinasas intracelulares a los trifosfatos (TP) respectivos, los cuales son fracciones activas con vidas medias intracelulares más largas que apoyan la dosificación una vez al día (ver Farmacocinética, Eliminación). Lamivudina-TP y carbovir-TP (las formas activas trifosfato de lamivudina y abacavir), son sustratos de e inhibidores competitivos de la transcriptasa reversa (TR) del VIH. Sin embargo, su principal actividad antiviral se realiza mediante la incorporación de la forma monofosfato a la cadena de ADN viral, resultando en la finalización de la cadena. Los trifosfatos de abacavir y lamivudina muestran una afinidad significativamente menor por las ADN polimerasas de la célula huésped. Efectos fármacodinámicos: En un estudio aleatorizado, de rangos de dosis, los sujetos infectados con VIH-1 tratados con monoterapia con dolutegravir (ING111521), demostraron una actividad antiviral rápida y dependiente de la dosis, con disminuciones medias desde la basal hasta el día 11 en el ARN del VIH-1 de 1.5; 2.0, y 2.5 log10 para dolutegravir 2 mg, 10 mg, y 50 mg 1 vez al día, respectivamente. Esta respuesta antiviral se mantuvo durante 3 a 4 días después de la última dosis en el grupo de 50 mg. Actividad antiviral en cultivos celulares: Las células mononucleares de sangre periférica (CMSP) infectadas con la cepa BaL del VIH-1 o con la cepa NL432 del VIH-1, proporcionaron DTG IC50 de 0.51 nM y 0.53 nM, respectivamente, y los ensayos de células MT-4 con la cepa IIIB del VIH-1, resultaron en IC50 de 0.71 y 2.1 nM. Cuando dolutegravir fue evaluado en ensayos de CMSP contra un panel consistente en 24 aislados clínicos de VIH-1 [grupo M (clade A, B, C, D, E, F y G) y grupo O] y 3 aislados clínicos de VIH-2, la media geométrica de la IC50 fue de 0.20 nM, y los valores de IC50 variaron de 0.02 a 2.14 nM para el VIH-1, mientras que la media geométrica de la IC50 fue de 0.18 nM, y los valores de IC50 variaron de 0.09 a 0.61 nM para los aislados del VIH-2. Actividad antiviral en combinación con otros agentes antivirales: Ningún fármaco con actividad intrínseca contra el VIH-1 fue antagónico con dolutegravir (se realizaron valoraciones in vitro en formato de tablero de ajedrez en combinación con estavudina, abacavir, efavirenz, nevirapina, lopinavir, amprenavir, enfuvirtida, maraviroc, adefovir y raltegravir). Además, los antivirales sin actividad inherente contra VIH (ribavirina), no tuvieron un efecto aparente sobre la actividad de dolutegravir. La actividad antiviral de abacavir en cultivos celulares, no fue antagonizada cuando se combinó con los inhibidores nucleósidos de la transcriptasa reversa (INTR) didanosina, emtricitabina, lamivudina, estavudina, tenofovir, zalcitabina o zidovudina, el inhibidor no nucleósido de transcriptasa reversa (INNTR) nevirapina, o el inhibidor de proteasas (IP) amprenavir. No se apreció ningún efecto antagonista in vitro con lamivudina y otros antirretrovirales (fármacos analizados: abacavir, didanosina, nevirapina, zalcitabina,y zidovudina). Efecto del suero humano y de las proteínas del suero: La IC90 ajustada para las proteínas (PA-IC90) en CMSP para dolutegravir, se calculó en 64 ng/ml. La concentración mínima de dolutegravir para una dosis única de 50 mg en sujetos sin tratamiento previo con inhibidores de integrasas, fue de 1.20 g/ml, 19 veces más alta que la PA-IC90 calculada. Los estudios de unión a proteínas plasmáticas in vitro indican que abacavir se une de forma baja a moderada (~49%) a las proteínas plasmáticas de humanos a concentraciones terapéuticas. Lamivudina muestra una farmacocinética lineal en el rango de dosis terapéutico, y muestra una unión baja a proteínas plasmáticas (menor de 36%). Resistencia in vitro (dolutegravir): Aislados de VIH-1 de tipo salvaje: No se observaron virus altamente resistentes a dolutegravir durante el pasaje de 112 días de la cepa IIIB, con un cambio máximo de 4.1 veces (FC) observado para los pasajes de poblaciones de virus resistentes con sustituciones en las posiciones conservadas IN S153Y y S153F. El pasaje de la cepa VIH-1 de tipo salvaje NL432 en presencia de dolutegravir, seleccionó para sustituciones E92Q (virus con pasaje de la población F C=3.1) y G193E (virus con pasaje de la población FC=3.2) el Día 56. El pasaje adicional de los subtipos salvajes de virus B, C, y A/G en presencia de dolutegravir, seleccionó para R263K, G118R, y S153T. Resistencia in vivo (dolutegravir): pacientes naďve a inhibidores de integrasas: No se aislaron mutaciones resistentes a INI o resistencia emergente con el tratamiento al tratamiento de base con INTR con dolutegravir 50 mg 1 vez al día en los estudios en pacientes naďve a tratamiento (estudios SPRING-1, SPRING-2, SINGLE y FLAMINGO). En el estudio SAILING en pacientes con tratamiento previo (y naďve a integrasas) (n=354 en el grupo de dolutegravir), se observaron sustituciones de integrasas emergentes con el tratamiento en la semana 48 en 4 de 17 sujetos con falla virológica en el grupo de dolutegravir. De estos 4, 2 sujetos tenían una sustitución única de integrasas en R263K, con un FC de 1.93, 1 sujeto tenía una sustitución polimórfica de integrasas V151V/I, con un FC de 0.92, y 1 sujeto tenía mutaciones preexistentes de integrasas, y se asume que había recibido tratamiento previo con integrasas o que se había infectado con un virus resistente a integrasas en el momento de la transmisión (ver Estudios clínicos). Resistencia in vitro e in vivo (abacavir y lamivudina): Se han seleccionado aislados de VIH-1 resistentes a abacavir in vitro e in vivo, y se asocian con cambios genotípicos específicos en la región TR del codón (codones M184V, K65R, L74V y Y115F). Durante la selección in vitro de abacavir, la mutación M184V ocurrió primero y resultó en un aumento de cerca del doble de la IC50, por debajo del corte clínico de abacavir de 4,5 FC. El pasaje continuo a mayores concentraciones del fármaco, resultó en la selección de mutantes dobles de TR 65R/184V y 74V/184V o mutantes triples de TR 74V/115Y/184V. Dos mutaciones confirieron una susceptibilidad a abacavir de 7 a 8 FC, y se requirieron combinaciones de 3 mutaciones para conferir una susceptibilidad de más de 8 FC. La resistencia del VIH-1 a lamivudina involucra el desarrollo de un cambio de aminoácidos M184I o M184V cercano al sitio activo de la TR viral. Esta variante surge tanto in vitro como en pacientes infectados con VIH-1 tratados con terapia antirretroviral que contiene lamivudina. Las mutantes M184V muestran una susceptibilidad a lamivudina mucho menor, así como una menor capacidad replicativa in vitro. M184V se asocia con un aumento de bajo nivel en la resistencia a abacavir, pero no confiere resistencia clínica a abacavir. Los aislados resistentes a abacavir también pueden mostrar menor sensibilidad a lamivudina. La combinación de abacavir/lamivudina ha demostrado una menor susceptibilidad a virus con sustituciones K65R con o sin la sustitución M184V/I, y a virus con L74V más la sustitución M184V/I. Efectos sobre el electrocardiograma: En un estudio aleatorizado, controlado con placebo, entrecruzado, 42 sujetos sanos recibieron la administraciones oral de dosis únicas de placebo, suspensión de dolutegravir de 250 mg (exposiciones aproximadamente 3 veces más altas con la dosis de 50 mg 1 vez al día en estado de equilibrio), y moxifloxacino (400 mg, control activo) en una secuencia aleatoria. Dolutegravir no aumentó el intervalo QTc 24 horas post dosis. Después del ajuste para la basal y para placebo, la media máxima del cambio del QTc en base al método de corrección de Fridericia (QTcF) fue de 1.99 mseg (IC unilateral superior del 95%: 4.53 mseg). No se realizaron estudios similares con abacavir ni lamivudina. Efectos sobre la función renal: Se evaluó el efecto de dolutegravir sobre la clearance de creatinina en suero (ClCr), la tasa de filtración glomerular (TFG) utilizando iohexol como sustrato, y el flujo plasmático renal efectivo (FPRE) utilizando para-aminohipurato (PAH) como sustrato, según el estudio abierto, aleatorizado, de 3 grupos, paralelo, controlado con placebo, en 37 sujetos sanos, quienes recibieron dolutegravir 50 mg 1 vez al día (n=12), 50 mg 2 veces al día (n=13) o placebo 1 vez al día (n=12) durante 14 días. Se observó una disminución modesta de la ClCr con dolutegravir en la primera semana de tratamiento, consistente con lo observado en los estudios clínicos. Dolutegravir con ambas dosis, no tuvo un efecto significativo sobre la TFG ni el FPRE. Estos datos apoyan los estudios in vitro que sugieren que los pequeńos aumentos de creatinina observados en los estudios clínicos se deben a la inhibición no patológica del transportador de cationes orgánicos 2 (OCT2) en los túbulos renales proximales, que median la secreción tubular de creatinina. Farmacocinética: El comprimido de Triumeq ha demostrado ser bioequivalente a Tivicay con Kivexa comprimidos administrados por separado. Esto se demostró en un estudio de bioequivalencia con dosis únicas, de 2 vías entrecruzadas, de Triumeq (en ayuno) versus 1 comprimido x 50 mg de dolutegravir, más 1 comprimido x 600 mg de abacavir/comprimido de 300 mg de lamivudina (en ayuno) en sujetos sanos (n=62). En una cohorte por separado, no se observó un efecto clínicamente significativo de una comida alta en grasa sobre la exposición a dolutegravir, abacavir o lamivudina. Éstos resultados indican que Triumeq puede tomarse con o sin alimentos. Las propiedades farmacocinéticas de dolutegravir, lamivudina y abacavir se describen abajo. Absorción: Dolutegravir, abacavir y lamivudina se absorben rápidamente después de la administración oral. No se ha establecido la biodisponibilidad absoluta de dolutegravir. La biodisponibilidad absoluta de abacavir oral y lamivudina en adultos es de 83% y 80 a 85% respectivamente. El tiempo medio hasta las concentraciones séricas máximas (tmáx) es de aproximadamente 2 a 3 horas (post-dosis para la fórmula en comprimido) para dolutegravir, 1.5 horas para abacavir y 1.0 horas para lamivudina. Después de dosis orales múltiples de dolutegravir 50 mg 1 vez al día, los cálculos de la media geométrica de los parámetros farmacocinéticos en estado de equilibrio son de 53.6 microgramos.h/ml para el AUC24, 3.67 microgramos/ml para la Cmáx, y de 1.11 microgramos/ml para la C24. Después de una dosis oral única de 600 mg de abacavir, la media de Cmáx es de 4.26 microgramos/ml y la media del AUC∞ es de 11.95 microgramos.h/ml. Después de la administración de dosis orales múltiples de lamivudina 300 mg 1 vez al día durante 7 días, la media en estado de equilibrio de la Cmáx es de 2.04 microgramos/ml, y la media del AUC24 es de 8.87 microgramos.h/ml. Distribución: El volumen de distribución aparente de dolutegravir (después de la administración oral de la fórmula en suspensión, Vd/F) se calculó en 12.5 L. Los estudios intravenosos con abacavir y lamivudina mostraron que la media del volumen de distribución aparente es de 0.8 y 1.3 l/kg, respectivamente. Dolutegravir se une en un alto grado a las proteínas plasmáticas de humanos (aproximadamente 99.3%) en base a los datos in vitro. La unión de dolutegravir a las proteínas plasmáticas fue independiente de la concentración. Las relaciones de la concentración de radioactividad relacionada con el fármaco en sangre total y en plasma, promediaron 0.441 a 0.535, indicando una asociación mínima de la radioactividad con los componentes celulares de la sangre. La fracción libre de dolutegravir en plasma se calcula en aproximadamente 0.2 a 1.1% en sujetos sanos, aproximadamente 0.4 a 0.5% en sujetos con insuficiencia hepática moderada, y 0.8 a 1.0% en sujetos con insuficiencia renal severa, y 0.5% en pacientes infectados con VIH-1. Los estudios de unión a proteínas plasmáticas in vitro indican que abacavir se une solamente de forma baja a moderada (aproximadamente 49%) a las proteínas plasmáticas de humanos a las concentraciones terapéuticas. Lamivudina muestra una farmacocinética lineal en el rango de dosis terapéutica, y muestra una unión baja de proteínas plasmáticas (menor de 36%). Dolutegravir, abacavir y lamivudina están presentes en el líquido cefalorraquídeo (LCR). En 12 sujetos naďve a tratamiento que recibieron un régimen de dolutegravir más abacavir/lamivudina durante 16 semanas, la concentración de dolutegravir en LCR promedió 15.4 ng/ml en la semana 2 y 12.6 ng/ml en la semana 16, variando de 3.7 a 23.2 ng/ml (comparable a la concentración plasmática no unida). La relación de la concentración en LCR: plasma de DTG, varió de 0.11 a 2.04%. Las concentraciones de dolutegravir en LCR superaron la IC50, apoyando la mediana de reducción desde la basal en el ARN de VIH-1 en el LCR de 2.2 log después de 2 semanas, y de 3.4 log después de 16 semanas de tratamiento (ver Farmacodinámica). Los estudios con abacavir demuestran una relación del AUC en LCR a plasma de entre 30 a 44%. Los valores observados de las concentraciones máximas son 9 veces mayores que la IC50 de abacavir de 0.08 microgramos/ml o 0.26 micromolar cuando abacavir se administra con dosis de 600 mg 2 veces al día. La media de la relación de las concentraciones de lamivudina en LCR/suero de 2 a 4 h después de la administración oral, fue de aproximadamente 12%. Se desconoce el grado real de penetración al SNC de lamivudina y su relación con la eficacia clínica. Dolutegravir está presente en el tracto genital de hombres y mujeres. El AUC en líquido cervicovaginal, tejido cervical, y tejido vaginal, fue de 6 a 10% del correspondiente observado en plasma en estado de equilibrio. El AUC fue de 7% en semen y de 17% en tejido rectal, del correspondiente observado en plasma en estado de equilibrio. Metabolismo: Dolutegravir se metaboliza principalmente mediante UGT1A1 con un componente menor del CYP3A (9.7% de la dosis total administrada en un estudio de balance de masas en humanos). Dolutegravir es el principal compuesto circulante en el plasma; la eliminación renal del fármaco sin cambios es baja (<1% de la dosis). Cincuenta y tres por ciento de la dosis oral total se excreta sin cambios en las heces. Se desconoce si todo o parte de esto se debe al fármaco no absorbido o a la excreción biliar del conjugado glucoronado, que puede ser degradado posteriormente para formar el compuesto principal en la luz del intestino. Treinta y uno por ciento de la dosis oral total se excreta en la orina, representada por el glucurónido de dolutegravir (18.9% de la dosis total), el metabolito de N-dealquilación (3.6% de la dosis total), y el metabolito formado por oxidación en el carbono bencílico (3.0% de la dosis total). Abacavir se metaboliza principalmente en el hígado, y menos del 2% de la dosis administrada se excreta por vía renal como compuesto sin cambios. Las principales vías de metabolismo en hombres son mediante el alcohol deshidrogenasa y mediante glucuronidación, para producir el 5'-ácido carboxílico y el 5'-glucurónido, que representan cerca del 66% de la dosis administrada. Estos metabolitos se excretan en la orina. El metabolismo de lamivudina es una vía menor de eliminación. Lamivudina se elimina principalmente sin cambios mediante excreción renal. La probabilidad de interacciones metabólicas con lamivudina es baja debido al bajo grado de metabolismo hepático (menor de 10%). Eliminación: Dolutegravir tiene una vida media terminal de ~14 horas, y una eliminación aparente (CL/F) de 0.56 l/hr. La media de la vida media de abacavir es de aproximadamente 1.5 horas. La media geométrica de la vida media terminal de carbovir-TP intracelular en estado de equilibrio es de 20.6 horas. Después de dosis orales múltiples de abacavir 300 mg 2 veces al día, no se observa una acumulación significativa de abacavir. La eliminación de abacavir se realiza mediante metabolismo hepático, con excreción subsecuente de los metabolitos principalmente en la orina. Los metabolitos y abacavir sin cambios representan aproximadamente 83% de la dosis administrada de abacavir en la orina. El resto se elimina en las heces. La vida media observada de la eliminación de lamivudina es de 5 a 7 horas. En pacientes que reciben lamivudina 300 mg 1 vez al día, la vida media terminal intracelular de lamivudina-TP aumentó a 16-19 horas. La media de eliminación sistémica de lamivudina es de aproximadamente 0.32 l/h/kg, principalmente mediante eliminación renal (mayor de 70%) mediante el sistema de transporte de cationes orgánicos. Poblaciones especiales de pacientes: Nińos: En un estudio pediátrico que incluyó 23 nińos y adolescentes infectados con VIH-1 con tratamiento antirretroviral previo, de 12 a 18 ańos de edad, se evaluó la farmacocinética de dolutegravir en 10 nińos, y mostró que la dosis de dolutegravir 50 mg 1 vez al día, resultó en una exposición de dolutegravir en sujetos pediátricos comparable a la observada en adultos que recibieron dolutegravir 50 mg 1 vez al día (Tabla 6).
Están disponibles datos limitados en adolescentes que recibieron una dosis diaria de 600 mg de abacavir y de 300 mg de lamivudina. Los parámetros farmacocinéticos son comparables a los reportados en adultos. Ancianos: El análisis farmacocinético poblacional de dolutegravir utilizando los datos en adultos infectados con VIH-1, mostró que no se observó un efecto clínicamente relevante de la edad sobre la exposición a dolutegravir. Los datos farmacocinéticos de dolutegravir, abacavir y lamivudina en sujetos >65 ańos de edad son limitados. Insuficiencia hepática: Se han obtenido los datos farmacocinéticos de dolutegravir, abacavir y lamivudina solos. En base a los datos obtenidos para abacavir, no se recomienda el uso de Triumeq en pacientes con insuficiencia hepática moderada y severa. Abacavir se metaboliza principalmente en el hígado. Se ha estudiado la farmacocinética de abacavir en pacientes con insuficiencia hepática leve (puntuación Child-Pugh 5 a 6). Los resultados mostraron que se observó un aumento medio de 1.89 veces en el AUC de abacavir y de 1.58 veces en la vida media de abacavir. Las AUC de los metabolitos no se vieron modificadas por la enfermedad hepática. Sin embargo, los índices de formación y eliminación de estos disminuyeron. Puede ser necesario disminuir la dosis de abacavir en pacientes con insuficiencia hepática leve. Por lo tanto, puede utilizarse la preparación por separado de Ziagen para tratar a estos pacientes. No se ha estudiado la farmacocinética de abacavir en pacientes con insuficiencia hepática moderada o severa. Se espera que las concentraciones plasmáticas de abacavir sean variables y aumenten sustancialmente en estos pacientes. Por lo tanto, no se recomienda el uso de Triumeq en pacientes con insuficiencia hepática moderada y severa. Los datos obtenidos con lamivudina en pacientes con insuficiencia hepática moderada y severa, y con dolutegravir en pacientes con insuficiencia hepática moderada, muestran que la farmacocinética no se ve afectada de forma significativa por la disfunción hepática. Dolutegravir se metaboliza y elimina principalmente en el hígado. En un estudio que comparó a 8 sujetos con insuficiencia hepática moderada (Child-Pugh categoría B) con 8 controles adultos sanos idénticos, la exposición a una dosis única de 50 mg de dolutegravir fue similar entre ambos grupos. No se ha estudiado el efecto de la insuficiencia hepática severa sobre la farmacocinética de dolutegravir. Insuficiencia renal: Se han obtenido datos farmacocinéticos de dolutegravir, abacavir y lamivudina solos. Triumeq no debe utilizarse en pacientes con una clereance de creatinina menor de 50 ml/min, porque a pesar de que no es necesario ajustar la dosis de dolutegravir ni de abacavir en pacientes con insuficiencia renal, se requiere disminuir la dosis del componente lamivudina. Por lo tanto, debe utilizarse la preparación por separado de Epivir para tratar a estos pacientes. Los estudios con lamivudina muestran que las concentraciones plasmáticas (AUC) aumentan en pacientes con disfunción renal debido a una disminución de la eliminación. Abacavir se metaboliza principalmente en el hígado, y aproximadamente 2% de abacavir se excreta sin cambios en la orina. La farmacocinética de abacavir en pacientes con enfermedad renal en estadio final es similar a la de los pacientes con función renal normal. La eliminación renal del fármaco sin cambios es una vía menor de eliminación de dolutegravir. Se realizó 1 estudio de la farmacocinética de dolutegravir en sujetos con insuficiencia renal severa (CLcr <30 ml/min). No se observaron diferencias farmacocinéticas clínicamente importantes entre sujetos con insuficiencia renal severa (CLcr <30 ml/min) y sujetos sanos idénticos. Dolutegravir no ha sido estudiado en pacientes con diálisis, aunque no se esperan diferencias en la exposición. Polimorfismos de las enzimas metabolizadoras de fármacos: No existe evidencia que indique que los polimorfismos comunes de las enzimas metabolizadoras de fármacos, alteren la farmacocinética de dolutegravir en un grado clínicamente relevante. En un meta análisis que utilizó muestras farmacogenómicas recolectadas en los estudios clínicos en sujetos sanos, los sujetos con genotipos UGT1A1 (n=7) que implican un mal metabolismo de dolutegravir, presentaron una eliminación 32% menor de dolutegravir y un AUC 46% mayor en comparación con sujetos con genotipos asociados con metabolismo normal mediante UGT1A1 (n=41). Los polimorfismos de CYP3A4, CYP3A5, y NR1I2 no se asociaron con diferencias en la farmacocinética de dolutegravir. Género: La exposición a dolutegravir en sujetos sanos parece ser ligeramente más alta (~20%) en mujeres que en hombres, en base a los datos obtenidos en un estudio en sujetos sanos (hombres n=17, mujeres n=24). Los análisis PK poblacionales que utilizaron datos farmacocinéticos agrupados de los estudios en adultos de Fase IIb y Fase III, no mostraron un efecto clínicamente relevante del género sobre la exposición a dolutegravir. No existe evidencia que indique que se requiere un ajuste de la dosis de dolutegravir, abacavir o lamivudina en base a los efectos del género sobre los parámetros PK. Raza: Los análisis PK poblacionales que utilizaron los datos farmacocinéticos agrupados de los estudios en adultos de Fase IIb y Fase III, no mostraron un efecto clínicamente relevante de la raza sobre la exposición a dolutegravir. La farmacocinética de dolutegravir después de la administración oral de dosis únicas en sujetos japoneses, parece ser similar a los parámetros observados en sujetos occidentales (Estados Unidos). No existe evidencia que indique que se requiere un ajuste de la dosis de dolutegravir, abacavir o lamivudina en base a los efectos de la raza sobre los parámetros PK. Coinfección con hepatitis B o C: Los análisis PK poblacionales indicaron que la coinfección por el virus de la hepatitis C no tuvo un efecto clínicamente relevante sobre la exposición a dolutegravir. Existen datos farmacocinéticos limitados en sujetos con coinfección por hepatitis B (ver Advertencias y Precauciones para el uso de Triumeq en pacientes co infectados con hepatitis B). Estudios clínicos: Sujetos sin tratamiento previo (naďve) con antirretrovirales: La eficacia de Triumeq en sujetos infectados con VIH, naďve a tratamiento antirretroviral, se basa en los análisis de los datos de 3 estudios; SINGLE (ING114467), SPRING-2 (ING113086) y FLAMINGO (ING114915). En SINGLE, 833 sujetos fueron aleatorizados, y recibieron al menos una dosis de dolutegravir 50 mg 1 vez al día con dosis fijas de abacavir-lamivudina (DTG + ABC/3TC) o dosis fijas de efavirenz-tenofovir-emtricitabina (EFV/TDF/FTC). En la basal, la mediana de edad de los pacientes era de 35 ańos, 16% eran mujeres, 32% no eran blancos, 7% tenían co infección por hepatitis C y 4% eran Clase CDC C; estas características eran similares entre grupos de tratamiento. Los resultados virológicos (incluyendo los resultados por variable basal clave) se describen abajo.
En el análisis principal de 48 semanas en el estudio SINGLE, la proporción de pacientes con supresión virológica (ARN del VIH-1 <50 copias/ml) en el grupo de dolutegravir + ABC/3TC (88%), fue superior a la del grupo de EFV/TDF/FTC (81%), p=0.003, se observó una diferencia similar entre tratamientos en los sujetos en base al nivel basal del ARN del VIH (< o > 100.000 copias/ml). La mediana de tiempo hasta la supresión viral fue de 28 días en el grupo que recibió dolutegravir + ABC/3TC y de 84 días en el grupo de EFV/TDF/FTC (p<0.0001). El cambio medio ajustado en el conteo de células T CD4+ desde la basal fue de 267 cels/mm3 en el grupo que recibió dolutegravir + ABC/3TC y de 208 cels/mm3 en el grupo de EFV/TDF/FTC en SINGLE a las 48 semanas [diferencia ajustada entre grupos (con un IC del 95%), 58.9 cels (33.4 cels a 84.4 cels), p<0.001]. Los análisis tanto del tiempo hasta la supresión viral como del cambio desde la basal, fueron preespecificados y ajustados para multiplicidad. A las 96 semanas, 80% de los participantes del estudio con el régimen de DTG + ABC/3TC, presentaban supresión virológica (<50 copias/ml) vs. 72% de los participantes en (EFV/TDF/FTC) [diferencia e IC del 95%; 8.0% (+2.3% a +13.8%); la diferencia en el criterio de valoración continuó siendo estadísticamente significativa, [p=0.006]. Las respuestas estadísticamente más altas en el grupo de DTG+ABC/3TC se debieron a retiros por EA, independientemente del estrato de la carga viral. A las 144 semanas en la fase abierta, se mantuvo la supresión virológica, el grupo de dolutegravir + ABC/3TC (71%) fue superior al grupo de EFV/TDF/FTC (63%), la diferencia entre los tratamientos fue 8.3 (2.0; 14.6). En SPRING-2, 822 adultos fueron aleatorizados y recibieron al menos una dosis de dolutegravir 50 mg 1 vez al día o raltegravir 400 mg 2 veces al día, ambos administrados como un tratamiento dual con dosis fijas de INTR (ABC/3TC o TDF/FTC). De estos sujetos, 169/411 en el grupo que recibió dolutegravir y 164/411 en el grupo que recibió raltegravir, estaban recibiendo régimen de base con ABC/3TC. En la basal, la mediana de la edad de los pacientes era de 36 ańos, 14% eran mujeres, 15% no eran blancos, 12% tenían coinfección con hepatitis B y/o C, y 2% eran CDC Clase C; estas características fueron similares entre grupos de tratamiento. La supresión virológica general [(ARN del VIH-1 <50 copias/ml) observada con el régimen de base en el grupo de dolutegravir (88%), no fue inferior a la del grupo de raltegravir (85%) a las 48 semanas. La diferencia ajustada en la proporción y el IC del 95% fueron de 2.5 (-2.2; 7.1)]. A las 96 semanas, la supresión virológica en el grupo de dolutegravir (81%) aún fue no inferior a la del grupo de raltegravir (76%). La diferencia ajustada en la proporción y el IC del 95% fue de 4.5 (-1.1; 10.0). Los índices de respuesta a las 48 semanas (y 96 semanas) fueron de 86% (y 74%) para dolutegravir + ABC/3TC y de 87% (y 76%) para raltegravir + ABC/3TC, respectivamente. En los estudios SINGLE y SPRING-2 de supresión virológica (ARN del VIH-1 <50 copias/ml), las diferencias entre tratamientos fueron comparables entre características basales (género, raza y edad). Durante las 96 semanas en SINGLE y SPRING-2, no se aislaron mutaciones de resistencia a INI ni de resistencia emergente con el tratamiento en los grupos que contenían dolutegravir. En SPRING-2, 4 sujetos del grupo de raltegravir, fallaron con mutaciones grandes de INTR, y un sujeto desarrolló resistencia a raltegravir; en SINGLE, 6 sujetos del grupo de EFV/TDF/FTC fallaron, con mutaciones asociadas con resistencia a INNTR y 1 desarrolló una mutación mayor de INTR. En FLAMINGO, un estudio abierto y controlado con activo, 485 adultos naďve a antirretrovirales infectados con VIH-1, fueron aleatorizados y recibieron al menos una dosis de dolutegravir 50 mg 1 vez al día o darunavir/ritonavir (DRV/r) 800 mg/100 mg 1 vez al día, ambos administrados como un tratamiento dual con dosis fijas de INTR (ABC/3TC o TDF/FTC). De estos sujetos, 33% en ambos grupos estaban recibiendo un régimen de base con ABC/3TC. En la basal, la mediana de edad de los pacientes era de 34 ańos, 15% eran mujeres, 28% no eran blancos, 10% tenían coinfección con hepatitis B y/o C, y 3% eran CDC Clase C; estas características fueron similares entre grupos de tratamiento. En general, la supresión virológica (ARN del VIH-1 <50 copias/ml) en el grupo de dolutegravir (90%) fue superior a la del grupo de DRV/r (83%) a las 48 semanas. La diferencia ajustada en la proporción del IC del 95% fue de 7.1 (+0.9 +13.2) [p=0,025]. A las 96 semanas la supresión virológica en el grupo de dolutegravir (80%) fue superior al grupo de DRV/r (68%). La mediana de tiempo hasta la supresión virológica fue de 28 días en el grupo de tratamiento con DTG y de 85 días en el grupo de DRV/r (p<0.001). Los índices de respuesta a las 48 semanas fueron de 90% para dolutegravir + ABC/3TC y de 85% para DRV/r/ABC/3TC y a las 96 semanas fue de un 82% para dolutegravir + ABC/3TC y de 75% para DRV/r/ABC/3TC. Ningún sujeto en el estudio presentó mutaciones de resistencia primaria emergentes con el tratamiento. Se demostró una respuesta virológica sostenida en el estudio SPRING-1 (ING112276), en el cual 88% de los pacientes que recibieron dolutegravir 50 mg (n=51) 1 vez al día presentaron ARN del VIH-1 <50 copias/ml, en comparación con 72% de los pacientes en el grupo de efavirenz (n=50) a las 96 semanas. Sujetos femeninos naďve a antirretrovirales: En ARIA (ING117172), un estudio aleatorio, abierto, con control activo, multicéntrico, de grupo paralelo, de no inferioridad; 499 mujeres adultas naďve a antirretrovirales infectadas con VIH-1 fueron asignadas al azar 1:1 para recibir cualquiera de las 2; DTG/ABC/3TC FDC 50 mg/600 mg/300 mg; O atazanamir 300 mg más ritonavir 100 mg más fumarato de disoproxilo de tenofovir/emtricitabina 300 mg/200 mg (ATV+RTV+TDF/FTC FCD), todos ellos administrados 1 vez al día. Las características demográficas fueron similares entre los grupos de tratamiento, al inicio la edad mediana de las pacientes fue de 37 ańos, el 45% de raza blanca y el 42% afroamericanas y africanas, el 93% resultó negativo para la hepatitis C (HCV) y el 84% de los sujetos eran clase A de la clasificación CDC. A las 48 semanas la supresión virológica general (ARN del VIH-1 <50 copias/ml) en el grupo DTG/ABC/3TC FDC (82%) demostró que era estadísticamente superior al grupo ATV + RTV + TDF/FTC FDC (71%). La diferencia ajustada en la proporción y IC del 95% fue de 10.5 (3.1% a 17.8%) [p=0.005]. No se aislaron mutaciones de resistencia a INI ni de resistencia emergente con el tratamiento con dolutegravir 50 mg 1 vez al día durante las 96 semanas. Sujetos con tratamiento previo con antirretrovirales: La eficacia de Triumeq también está apoyada por los datos de un estudio aleatorizado, internacional, doble ciego, controlado con activo, SAILING (ING111762). En el estudio SAILING, 719 adultos infectados con VIH-1, con tratamiento previo con TAR, naďve a inhibidores de integrasas fueron aleatorizados y recibieron dolutegravir 50 mg 1 vez al día o raltegravir 400 mg 2 veces al día con el régimen de base (RB) seleccionado por el investigador, consistente en 2 agentes (incluyendo al menos un agente totalmente activo). En la basal, la mediana de edad de los pacientes fue de 43 ańos, 32% eran mujeres, 50% no eran blancos, 16% tenían coinfección por hepatitis B y/o C, y 46% eran CDC Clase C. Todos los sujetos tenían al menos 2 clases de resistencia a TAR, y 49% de los sujetos tenían al menos 3 clases de resistencia a TAR en la basal. La supresión virológica (ARN del VIH-1 <50 copias/ml) en el grupo de dolutegravir (71%), fue estadísticamente superior a la del grupo de raltegravir (64%) en la Semana 48 (p=0.030). Las diferencias entre tratamientos en cuanto a la supresión virológica (ARN del VIH-1 <50 copias/ml) fueron comparables entre las características basales de género, raza y subtipo de VIH. En STRIIVING (201147) un estudio de 48 semanas de duración, aleatorizado, abierto, con control activo, multicéntrico, de no inferioridad; se asignaron al azar a 555 sujetos (1:1) infectados con VIH-1, virológicamente suprimidos (RNA del HIV-1 <50 c /ml) para continuar con su régimen actual de TAR (2 INTRs más un PI, INNTR o INI) o cambiar a ABC / DTG / 3TC FDC 1 vez al día (cambio temprano /Early Switch). La mayoría de los sujetos en la población por intención de tratar expuesta (ITT-E) eran blancos (65%) y hombres (86%); La mediana de edad fue de 45 (rango 22-80) ańos. En el basal, el 31% de los sujetos tenían recuentos de CD4 + de <500 células/mm3. En general, la mayoría de los sujetos presentaron resultados negativos en la detección de la infección por VHB y VHC (93%), eran clase A de la clasificación CDC (73%) e identificaron la actividad homosexual como factor de riesgo del VIH (72%). La supresión virológica (ARN del VIH-1 <50 copias/ml) en el grupo ABC / DTG / 3TC FDC (85%) fue estadísticamente no inferior a los grupos que mantuvieron su TAR (88%) a las 24 semanas. La diferencia ajustada en la proporción del CI del 95% [ABC / DTG / 3TC vs ART actual] fue del 3.4%; CI del 95%: [-9.1, 2.4]. Después de 24 semanas todos los sujetos restantes cambiaron a ABC / DTG / 3TC FDC (cambio tardío/Late Switch). Se mantuvieron niveles similares de supresión virológica tanto en los grupos de cambio temprano como tardío a las 48 semanas. En CAL30001 y ESS30008, ABC/3TC y ABC + 3TC se utilizaron de forma efectiva como tratamiento combinado para mantener la supresión viral en sujetos con tratamiento previo, con índices bajos de mutaciones de resistencia viral emergentes con el tratamiento. Nińos: En el estudio abierto de Fase I/II de 48 semanas, multicéntrico (P1093/ING112578), se evaluaron los parámetros farmacocinéticos, seguridad, tolerabilidad y eficacia de dolutegravir en regímenes combinados en lactantes, nińos y adolescentes infectados con VIH-1. A las 24 semanas, 16 de 23 (69%) adolescentes (12 a menos de 18 ańos de edad) tratados con dolutegravir 1 vez al día (35 mg n=4, 50 mg n=19) más régimen de base optimiado, lograron una carga viral menor de 50 copias/ml. Datos preclínicos de seguridad: Con la excepción de una prueba negativa de micronúcleos de ratas in vivo para la combinación de abacavir y lamivudina, no existen datos disponibles sobre los efectos de la combinación de dolutegravir, abacavir y lamivudina en animales.
Posología: Antes de iniciar el tratamiento con medicamentos que contengan abacavir, se debe llevar a cabo una prueba de detección del alelo HLA-B*5701 en los pacientes infectados por el VIH, independiente del origen racial. No se debe emplear abacavir en pacientes portadores del alelo HLA-B*5701. El tratamiento con Triumeq solo debe ser iniciado por un médico experimentado en el manejo de la infección por VIH. Triumeq no debe administrarse a adultos o adolescentes que pesen menos de 40 kg, debido a que es un comprimido de dosis fija y no puede reducirse la dosis. Triumeq puede tomarse con o sin alimentos. Triumeq es un comprimido de dosis fija y no debe ser prescrito en pacientes que requieran ajustes de la dosis, como aquellos con un clearance de creatinina menor de 50 ml/min. Las preparaciones por separado de dolutegravir, abacavir o lamivudina deben administrarse en casos en los que está indicada la suspensión o el ajuste de la dosis. En estos casos, el médico debe remitirse a la información individual del producto para estos medicamentos. Debido a que la dosis recomendada de dolutegravir es de 50 mg 2 veces al día en pacientes con resistencia a los inhibidores de integrasas, no se recomienda el uso de dolutegravir en pacientes con resistencia a los inhibidores de integrasas. Poblaciones: Adultos y adolescentes: La dosis recomendada de Triumeq en adultos y adolescentes que pesan al menos 40 kg es de 1 comprimido 1 vez al día. Dosis olvidadas: Si el paciente olvida tomar una dosis deTriumeq, debe tomar Triumeq tan pronto como sea posible, siempre y cuando la siguiente toma no sea antes de 4 horas. Si la siguiente toma es antes de 4 horas, el paciente no debe tomar la dosis olvidada y simplemente debe reanudar la pauta de dosificación habitual. Nińos: Actualmente, Triumeq no está recomendado para el tratamiento de nińos menores de 12 ańos de edad, ya que no es posible ajustar la dosis. Actualmente no existen datos clínicos disponibles para esta combinación. Los médicos deben ver la información individual del producto para dolutegravir, abacavir y lamivudina. Ancianos: Existen datos limitados disponibles sobre el uso de dolutegravir, abacavir y lamivudina en pacientes de 65 ańos y mayores. Sin embargo, no existe evidencia de que los pacientes ancianos requieran una dosis distinta en comparación con pacientes adultos más jóvenes (ver Farmacocinética - Poblaciones especiales de pacientes). Al tratar pacientes ancianos, debe tomarse en cuenta la frecuencia más alta de disminución de la función hepática, renal y cardíaca, uso de productos medicinales o enfermedades concomitantes. Insuficiencia renal: Al tiempo que no es necesario ajustar la dosis de dolutegravir o abacavir en pacientes con insuficiencia renal, se requiere una disminución de la dosis de lamivudina debido a una menor eliminación. Por lo tanto, no se recomienda el uso de Triumeq en pacientes con un clereance de creatinina menor de 50 ml/min (ver Farmacocinética - Poblaciones especiales de pacientes). Insuficiencia hepática: Puede ser necesario disminuir la dosis de abacavir en pacientes con insuficiencia hepática leve (Child-Pugh grado A). Debido a que no es posible disminuir la dosis de Triumeq, deben utilizarse las preparaciones por separado de dolutegravir, abacavir y lamivudina cuando se considere necesario. Triumeq no se recomienda en pacientes con insuficiencia hepática moderada y severa (Child-Pugh grado B o C) (ver Farmacocinética-Poblaciones especiales de pacientes).
Modo de Empleo:Instrucciones para el uso/manejo: No existen requisitos especiales para el uso/manejo de este producto. No están disponibles todas las presentaciones en todos los países. Importante-Tarjeta de alerta: Triumeq (dolutegravir/abacavir/lamivudina) comprimidos recubiertos: Lleve siempre consigo esta tarjeta. Debido a que Triumeq contiene abacavir, algunos pacientes recibiendo Triumeq pueden desarrollar una reacción de hipersensibilidad (reacción alérgica grave) que puede ser potencialmente mortal si continua con el tratamiento con Triumeq. Contacte inmediatamente a su doctor para que le indique si debe o no dejar de tomar Triumeq si usted: 1) Presenta una erupción cutánea. 2) Presenta 1 o más síntomas de un mínimo de 2 de los siguientes grupos: fiebre; dificultad para respirar, dolor de garganta o tos; náusea o vómito o diarrea o dolor abdominal; cansancio severo o molestia o malestar general. Si usted ha discontinuado Triumeq debido a esta reacción, usted nunca deve volver a tomar Triumeq , o cualquier otro medicamento conteniendo abacavir (Ziagen, Kivexa o Tricivir) debido a que en horas puede volver a experimentar disminución potencialmente mortal de su presión arterial o incluso la muerte.
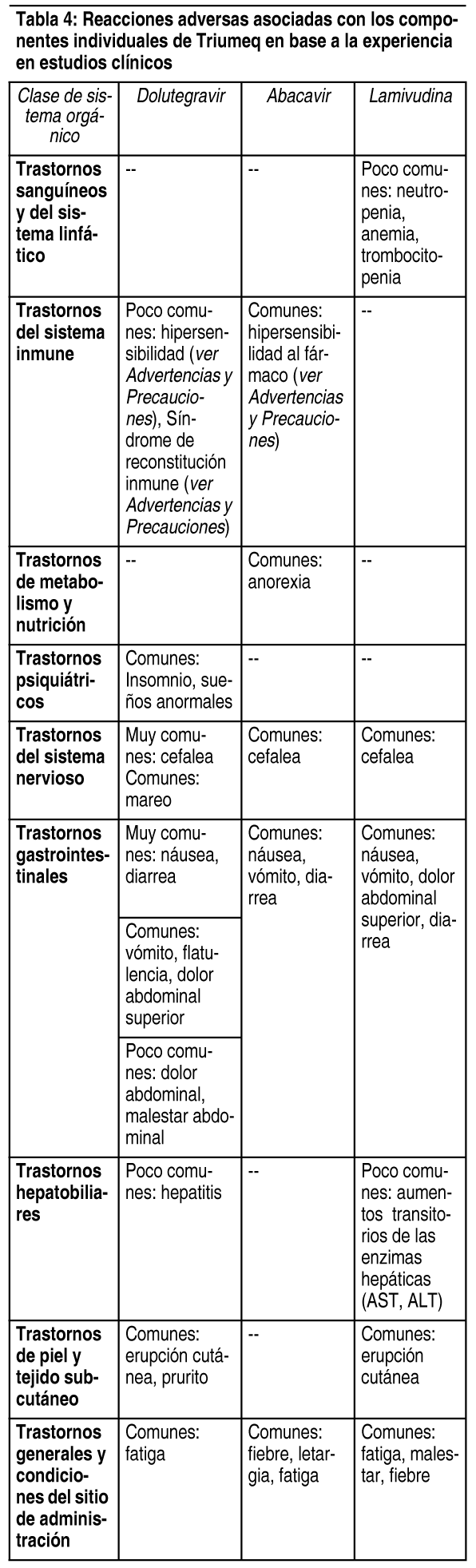
Efectos Colaterales: Triumeq contiene dolutegravir, abacavir y lamivudina, por lo que pueden esperarse los eventos adversos asociados con estos. Para muchos de los eventos adversos detallados, no está claro si están relacionados con la sustancia activa, el amplio rango de otros medicamentos utilizados para el manejo de la infección por VIH, o si son resultado del proceso de la enfermedad de base. Muchos de los eventos adversos detallados ocurren frecuentemente (náusea, vómito, diarrea, fiebre, letargia, exantema) en pacientes con hipersensibilidad a abacavir. Por lo tanto, los pacientes con cualquiera de estos síntomas deben ser evaluados cuidadosamente buscando la presencia de esta reacción de hipersensibilidad. Si se ha suspendido Triumeq en los pacientes debido a que experimentaron cualquiera de estos síntomas y se debe tomar la decisión de reiniciar abacavir, esto debe ser realizado sólo bajo supervisión médica directa (ver Consideraciones especiales después de interrumpir el tratamiento con Triumeq en Advertencias y precauciones). Las reacciones adversas al fármaco de dolutegravir, abacavir o lamivudina se detallan en las tablas que se encuentran abajo por clase de sistema orgánico MedDRA y por frecuencia. La frecuencia se define como: Muy comunes (≥1/10); Comunes (≥1/100, <1/10); Poco comunes (≥1/1.000, <1/100); Raras (≥1/10.000, <1/1.000); Muy raras (<1/10.000), incluyendo reportes aislados. Datos de estudios clínicos: Los datos de seguridad clínica con Triumeq son limitados. Las reacciones adversas observadas con la combinación de dolutegravir más abacavir/lamivudina en un análisis de los datos agrupados en los estudios clínicos de Fase IIb a Fase IIIb, en general fueron consistentes con los perfiles de reacciones adversas de los componentes individuales dolutegravir, abacavir y lamivudina. Sin embargo, se observaron las siguientes reacciones adversas comunes emergentes con el tratamiento con la combinación, que no estaban detalladas en la información de prescripción de ninguno de los componentes individuales: Trastornos gastrointestinales: distensión abdominal, enfermedad por reflujo gastroesofágico, dispepsia. Trastornos del sistema nervioso: somnolencia. Trastornos psiquiátricos: pesadillas y trastornos del sueńo. Trastornos de metabolismo y nutrición: hipertrigliceridemia e hiperglucemia. Además, se observaron fatiga e insomnio con una frecuencia más alta con dolutegravir más abacavir/lamivudina en comparación con los componentes individuales. La categoría de frecuencia de fatiga e insomnio fue 'muy común' con la combinación (previamente 'común' con cada componente individual o con dolutegravir, respectivamente). No se observaron diferencias entre la combinación y los componentes individuales en cuanto a la severidad de ninguna reacción adversa observada.
Cambios en las químicas de laboratorio: Ocurrieron aumentos de la creatinina sérica en la primera semana de tratamiento con dolutegravir, que se mantuvieron estables durante 96 semanas. En ING114467, se observó un cambio medio desde la basal de 12.6 µmol/l después de 96 semanas de tratamiento. Estos cambios no se consideran clínicamente relevantes, ya que no reflejan un cambio en la tasa de filtración glomerular (ver Farmacodinámica - Efectos sobre la función renal). Se observaron aumentos leves de la bilirrubina total (sin ictericia clínica) en los grupos de dolutegravir y raltegravir (pero no en los de efavirenz) en el programa. Estos cambios no se consideran clínicamente relevantes, ya que probablemente reflejan la competencia entre dolutegravir y la bilirrubina no conjugada por una vía de eliminación común (UGT1A1) (ver Farmacocinética - Metabolismo). También se han reportado aumentos aislados de la creatina fosfoquinasa (CPK) asociados principalmente con el ejercicio con el tratamiento con dolutegravir. Población pediátrica: No existen datos de estudios clínicos sobre los efectos de Triumeq en la población pediátrica. Los componentes individuales se han investigado en adolescentes de 12 a 18 ańos de edad. En base a los datos limitados disponibles sobre el uso de dolutegravir como entidad única combinado con otros agentes antirretrovirales para tratar adolescentes (12 a menores de 18 ańos de edad), no se observaron otros tipos adicionales de reacciones adversas distintos a los observados en la población adulta. Las preparaciones individuales de ABC y 3TC se han investigado por separado, y como tratamiento de base dual con nucleósidos, combinados con tratamiento antirretroviral para tratar pacientes pediátricos infectados con VIH sin tratamiento previo (naďve) conTAR o con tratamiento previo con TAR (los datos disponibles sobre el uso de ABC y 3TC en nińos menores de tres meses son limitados). No se han observado otros tipos de efectos indeseables más allá de los caracterizados en la población de adultos. Datos post-comercialización: Además de las reacciones adversas incluidas en los datos de los estudios clínicos, se han identificado las reacciones adversas detalladas en la Tabla 5 durante el uso post-aprobación de abacavir y lamivudina. Se han elegido estos eventos para ser incluidos debido a una potencial conexión causal con abacavir y/o lamivudina. No están disponibles datos post-comercialización sobre dolutegravir .
Se ha observado redistribución/acumulación de grasa corporal en algunos pacientes que reciben tratamiento antirretroviral combinado (ver Advertencias y precauciones). La incidencia de este evento depende de varios factores, incluyendo la combinación particular de fármacos antirretrovirales. Descripción de reacciones adversas seleccionadas: Hipersensibilidad (ver también Advertencias y precauciones): Tanto abacavir como dolutegravir se asocian a un riesgo de reacciones de hipersensibilidad (RHS), que se observaron más comúnmente con abacavir. La reacción de hipersensibilidad observada para cada uno de estos productos medicinales (descrita a continuación) comparte algunas características comunes como fiebre y/o erupción cutánea con otros síntomas que indican un involucramiento de múltiples órganos. El tiempo para el inicio fue típicamente 10-14 días tanto para las reacciones asociadas con abacavir como con dolutegravir, aunque las reacciones a abacavir pueden ocurrir en cualquier momento durante la terapia. Hipersensibilidad a dolutegravir: Los síntomas han incluido erupción cutánea, hallazgos constitucionales y algunas veces disfunción orgánica, incluyendo reacciones hepáticas severas. Hipersensibilidad a abacavir: A continuación se enlistan los signos y síntomas de esta reacción de hipersensibilidad (RHS). Éstos se han identificado ya sea a partir de estudios clínicos o de la vigilancia posterior a la comercialización. Aquellos reportados en al menos 10% de los pacientes con una reacción de hipersensibilidad se encuentran en texto en negritas. Casi todos los pacientes que desarrollan reacciones de hipersensibilidad tendrán fiebre y/o erupción cutánea (comúnmente maculopapular o urticariano) como parte del síndrome, no obstante, se han presentado reacciones sin erupción cutánea o fiebre. Otros síntomas clave incluyen síntomas gastrointestinales, respiratorios o constitucionales como letargo y malestar. Piel: Erupción cutánea (a menudo maculopapular o urticariano). Tracto gastrointestinal: Náusea, vómito, diarrea, dolor abdominal, ulceración bucal. Tracto respiratorio: Disnea, tos, dolor de garganta, síndrome de dificultad respiratoria en adultos, insuficiencia respiratoria. Misceláneos: Fiebre, fatiga, malestar, edema, linfadenopatía, hipotensión, conjuntivitis, anafilaxia. Neurológicos/psiquiátricos: Dolor de cabeza, parestesia. Hematológicos: Linfopenia. Hígado/páncreas: Pruebas de función hepática elevadas, insuficiencia hepática. Musculoesqueléticas: Mialgia, raras veces miolisis, artralgia, creatina fosfoquinasa elevada. Urología: Creatinina elevada, insuficiencia renal. Reiniciar abacavir después de una RHS a abacavir resulta en un rápido regreso de los síntomas en horas. Esta recurrencia de la RHS es a menudo más severa que en la presentación inicial y puede incluir hipotensión potencialmente mortal y muerte. También han ocurrido reacciones con poca frecuencia después de reiniciar abacavir en pacientes que tuvieron sólo uno de los síntomas clave de hipersensibilidad (ver arriba) antes de suspender abacavir; y en muy raras ocasiones también se han observado en pacientes que han reiniciado terapia sin síntomas precedentes de una RHS (es decir, pacientes considerados previamente tolerantes a abacavir). Para obtener detalles del control clínico en caso de una RHS sospechosa a abacavir ver Advertencias y precauciones.
Contraindicaciones: Triumeq está contraindicado en pacientes con hipersensibilidad conocida a dolutegravir, abacavir o lamivudina, o a cualquiera de los excipientes. Triumeq está contraindicado en combinación con dofetilida o pilsicainida.
Advertencias: Se incluyen en esta sección las advertencias y precauciones especiales relacionadas con dolutegravir, abacavir y lamivudina. No existen advertencias y precauciones adicionales relacionadas con Triumeq. Reacciones de hipersensibilidad (ver también Efectos colaterales): Tanto abacavir como dolutegravir se asocian a un riesgo de reacciones de hipersensibilidad (RHS) (ver Descripción clínica de RHS a continuación y Efectos colaterales) y comparten algunas características en común como fiebre y/o erupción cutánea con otros síntomas que indican una implicación de múltiples órganos. Clínicamente no es posible determinar si una RHS con Triumeq es causada por abacavir o dolutegravir. Se han observado RHS más comúnmente con abacavir, algunas de las cuales han sido potencialmente mortales y en raros casos fatales. El riesgo de que ocurra una RHS por abacavir aumenta significativamente en pacientes con resultados positivos en la prueba del alelo HLA-B*5701. No obstante, las RHS por abacavir han sido reportadas en pacientes que no poseen este alelo. Se debería cumplir con lo siguiente: La evaluación del estado de HLA-B*5701 debería considerarse antes de iniciar el tratamiento y también antes de reiniciar el tratamiento con abacavir en pacientes con estado HLA-B*5701 desconocido que toleraron el abacavir previamente. No se recomienda el uso de Triumeq en pacientes que presentan el alelo HLA-B*5701 ni en pacientes con sospecha previa de RHS por abacavir mientras tomaban cualquier otro medicamento con abacavir (por ej.: Ziagen, Kivexa, Tricivir) sin importar la condición de HLA-B*5701. Se le deberá recordar a cada paciente leer el folleto para el paciente incluido en el estuche de Triumeq. Se les debería recordar la importancia de tomar la tarjeta de alerta incluida en el empaque y conservarla en todo momento. En cualquier paciente tratado con Triumeq, el diagnóstico clínico de sospecha de reacción de hipersensibilidad debe ser la base de la toma de decisiones clínicas. Triumeq debe detenerse sin retraso, incluso en la ausencia del alelo HLA-B*5701, si se sospecha de RHS. El retraso para detener el tratamiento con Triumeq después del inicio de la hipersensibilidad podría resultar en una reacción de riesgo para la vida. Debe monitorearse el estado clínico incluyendo aminotransferasas hepáticas y bilirrubina. Se les deberá indicar a los pacientes que han experimentado reacciones de hipersensibilidad, eliminar sus comprimidos restantes de Triumeq con el fin de evitar reiniciar el tratamiento con abacavir. Después de detener el tratamiento con Triumeq por razones de una RHS sospechosa, jamás debe reiniciarse Triumeq o cualquier otro producto medicinal que contenga abacavir o dolutegravir. Reiniciar el tratamiento con productos que contienen abacavir después de una RHS sospechosa a abacavir, puede resultar en un reinicio rápido de los síntomas en horas y puede incluir hipotensión de riesgo para la vida y muerte. Si se descarta una reacción de hipersensibilidad, los pacientes pueden reiniciar el tratamiento con Triumeq. Pocas veces, los pacientes que detuvieron el tratamiento con abacavir por otras razones además de los síntomas de RHS también experimentaron reacciones que ponen en peligro su vida a horas de reiniciar el tratamiento con abacavir (ver la Sección Descripción de reacciones adversas seleccionadas). Los pacientes deben estar conscientes de que pueden ocurrir RHS al reiniciar Triciriv o cualquier otro medicamento que contenga abacavir (por ejemplo: Ziagen, Kivexa, Triumeq) y que reiniciar Tricivir o cualquier otro medicamento que contenga abacavir, debería asumirse sólo si puede accederse rápidamente a la atención médica. Descripción clínica de RHS por dolutegravir: Se han reportado reacciones de hipersensibilidad con los inhibidores de integrasa, incluyendo dolutegravir, las cuales se caracterizaron por erupción cutánea, hallazgos constitucionales y en ocasiones, disfunción orgánica, incluyendo lesión hepática. Descripción clínica de RHS por abacavir: Las RHS con abacavir han sido bien caracterizadas a través de estudios clínicos y durante seguimiento posterior a la comercialización. Los síntomas usualmente aparecen dentro de las primeras 6 semanas (mediana de tiempo de comienzo 11 días) de inicio del tratamiento con abacavir, aunque estas reacciones pueden ocurrir en cualquier momento del tratamiento. Casi todas las RHS por abacavir tienen fiebre y/o erupción cutánea como parte del síndrome. Otros signos y síntomas que se han observado como parte de las RHS por abacavir incluyen síntomas respiratorios y gastrointestinales, que podrían llevar a un diagnóstico equivocado de RHS como enfermedad respiratoria (neumonía, bronquitis, faringitis) o gastroenteritis (ver Efectos colaterales, Descripción de reacciones adversas seleccionadas). Los síntomas relacionados con RHS empeoran al continuar con la administración y se puede poner en peligro la vida del paciente. Generalmente, estos síntomas se resuelven al suspender la administración de abacavir. Acidosis láctica/hepatomegalia grave con esteatosis: La acidosis láctica y la hepatomegalia grave con esteatosis, incluyendo casos fatales, se han reportado con el uso de antirretrovirales análogos de los nucleósidos, ya sean solos o combinados, incluyendo abacavir y lamivudina. La mayoría de estos casos se han presentado en mujeres. Las características clínicas que pueden indicar el desarrollo de acidosis láctica, incluyen debilidad generalizada, anorexia, y pérdida súbita e inexplicable de peso, síntomas gastrointestinales y síntomas respiratorios (disnea y taquipnea). Debe tenerse precaución al administrar Triumeq, particularmente en aquellos con factores de riesgo conocidos para enfermedad hepática. Debe suspenderse el tratamiento con Triumeq en cualquier paciente que desarrolle hallazgos clínicos o de laboratorio sugestivos de acidosis láctica con o sin hepatitis (que pueden incluir hepatomegalia y esteatosis, incluso en ausencia de aumentos marcados de las transaminasas). Lipodistrofia: Se han observado por separado o juntos, redistribución/acumulación de la grasa corporal, incluyendo obesidad central, aumento de la grasa dorsocervical (joroba de búfalo), adelgazamiento periférico, adelgazamiento facial, aumento de las mamas, aumento de los niveles de lípidos en suero y de glucosa en sangre, en algunos pacientes que reciben tratamiento combinado con antirretrovirales (ver Efectos colaterales). Al tiempo que todos los miembros de las clases de medicamentos que son inhibidores de proteasas e inhibidores nucleósidos de la transcriptasa reversa, se han asociado con 1 o más de estos eventos adversos específicos, vinculados a un síndrome general comúnmente llamado lipodistrofia, los datos indican que existen diferencias en el riesgo entre miembros individuales de las clases terapéuticas respectivas. Además, el síndrome de lipodistrofia tiene una etiología multifactorial; por ejemplo estado de la enfermedad por VIH, edad más avanzada y mayor duración del tratamiento antirretroviral, todos juegan papeles importantes, posiblemente sinérgicos. Actualmente se desconocen las consecuencias a largo plazo de estos eventos. La exploración clínica debe incluir la evaluación de signos físicos de redistribución de la grasa. Debe tomarse en cuenta la medición de los lípidos en suero y de la glucosa en sangre. Los trastornos de lípidos deben manejarse según se considere clínicamente adecuado. Síndrome de reconstitución inmune: En pacientes infectados con HIV con deficiencia inmune grave en el momento de iniciar el tratamiento antirretroviral (TAR), puede surgir una reacción inflamatoria a infecciones asintomáticas u oportunistas residuales, y ocasionar condiciones clínicas graves o agravamiento de los síntomas. Típicamente, dichas reacciones se han observado en las primeras semanas o meses de iniciado el TAR. Los ejemplos relevantes son retinitis por citomegalovirus, infecciones generalizadas y/o focales por micobacterias y neumonía por Pneumocystis jiroveci (a menudo referico como PCP). Cualquier síntoma inflamatorio debe ser evaluado sin demora, e iniciar el tratamiento siempre que sea necesario. También se han reportado trastornos autoinmunes (como enfermedad de Graves, polimiositis y síndrome de Guillain-Barré) en el escenario de reconstitución inmune, sin embargo, el tiempo hasta el inicio es más variable, y puede ocurrir varios meses después de iniciado el tratamiento, y algunas veces puede tener una presentación atípica. Se observaron aumentos en las pruebas de función hepática consistentes con síndrome de reconstitución inmune en algunos pacientes co infectados con hepatitis B y/o C al iniciar el tratamiento con dolutegravir. Se recomienda el monitoreo de las pruebas de función hepática en pacientes con coinfección por hepatitis B y/o C (ver Pacientes coinfectados con virus de la hepatitis B (VHB) más adelante en esta sección y Efectos colaterales). Pacientes coinfectados con virus de la hepatitis B (VHB): Debe tenerse particular cuidado al iniciar o mantener un tratamiento eficaz contra la hepatitis B al iniciar el tratamiento con Triumeq en pacientes coinfectados con hepatitis B. Los estudios clínicos y el uso comercializado de lamivudina, han demostrado que algunos pacientes con enfermedad crónica por VHB pueden experimentar evidencia clínica o de laboratorio de hepatitis recurrente al suspender lamivudina, lo cual puede tener consecuencias más graves en pacientes con enfermedad hepática descompensada. Si Triumeq se suspende en pacientes coinfectados con HBV, debe considerarse el monitoreo periódico tanto de las pruebas de función hepática como de los marcadores de replicación del VHB. Infecciones oportunistas: Los pacientes que reciben Triumeq o cualquier otro tratamiento antirretroviral, aún pueden desarrollar infecciones oportunistas y otras complicaciones de la infección por VIH. Por lo tanto, los pacientes deben ser mantenidos bajo observación clínica estrecha por parte de médicos con experiencia en el tratamiento de estas enfermedades asociadas con el VIH. Transmisión de infecciones: Debe advertirse a los pacientes que el tratamiento antirretroviral actual, incluyendo Triumeq, no ha demostrado que prevenga el riesgo de transmisión del VIH a otros mediante contacto sexual o contaminación de la sangre. Deben continuar tomando las precauciones pertinentes. Infarto al miocardio: Varios estudios observacionales, epidemiológicos, han reportado una asociación entre el uso de abacavir y el riesgo de infarto al miocardio. En los metaanálisis de ensayos controlados aleatorizados no se ha observado mayor riesgo de infarto al miocardio con el uso de abacavir. Hasta la fecha no existe un mecanismo biológico establecido que explique un potencial aumento del riesgo. En su totalidad, los datos disponibles de los estudios observacionales y de los estudios clínicos controlados muestran inconsistencia y por lo tanto la evidencia de una relación causal entre el tratamiento con abacavir y el riesgo de infarto al miocardio no es concluyente. Como precaución, debe tomarse en cuenta el riesgo de base para enfermedad cardiaca coronaria al prescribir tratamientos antirretrovirales, incluyendo abacavir, y deben realizarse acciones que minimicen todos los factores de riesgo modificables (ej., hipertensión, hiperlipidemia, diabetes mellitus y tabaquismo). Interacciones farmacológicas: Debe tenerse precaución al coadministrar medicamentos (con y sin prescripción) que puedan cambiar la exposición a dolutegravir, abacavir, lamivudina o a medicamentos que vean afectada su exposición por Triumeq (ver Contraindicaciones e Interacciones). Triumeq no debe administrarse de forma concurrente con otros medicamentos que contengan cualquiera de los mismos componentes activos (dolutegravir, abacavir, y/o lamivudina). Debido a que la dosis recomendada de Tivicay es de 50 mg 2 veces al día en pacientes que toman etravirina (sin inhibidores de proteasa potenciados), efavirenz, nevirapina, rifampicina, tipranavir/ritonavir, carbamazepina, fenitoína, fenobarbital y hierba de San Juan. No se recomienda el uso de Triumeq en pacientes que toman estos medicamentos (ver Interacciones). Dolutegravir no debe coadministrarse con antiácidos que contienen cationes polivalentes. Se recomienda administrar Triumeq 2 horas antes ó 6 horas después de estos agentes (ver Interacciones). Se recomienda administrar Triumeq 2 horas antes ó 6 horas después de tomar suplementos de calcio o hierro, o de forma alterna, administrarlo con los alimentos (ver Interacciones). Dolutegravir incrementó las concentraciones de metformina. Se debe considerar un ajuste de la dosis de metformina cuando se inicie y suspenda la coadministración de dolutegravir con metformina, para mantener el control glicémico (ver Interacciones).
Precauciones:Embarazo y lactancia: Fertilidad: No existen datos sobre los efectos de dolutegravir, abacavir o lamivudina sobre la fertilidad en hombres o mujeres. Los estudios en animales no indican efectos de dolutegravir, abacavir o lamivudina sobre la fertilidad en machos o hembras (ver Datos preclínicos de seguridad). Embarazo: No se ha establecido la seguridad del uso de Triumeq sobre el embarazo en humanos. Se demostró que dolutegravir cruza la placenta en los estudios de toxicidad reproductiva en animales. Lamivudina y abacavir se asociaron con hallazgos en los estudios de toxicidad reproductiva en animales (ver Datos preclínicos de seguridad). Por lo tanto, la administración de Triumeq en el embarazo debe considerarse solo si el beneficio para la madre sobrepasa el posible riesgo para el feto. Se han realizado reportes de aumentos leves y transitorios en los niveles de lactato en suero, lo cual puede deberse a disfunción mitocondrial en neonatos y lactantes expuestos in utero o peri-parto a inhibidores nucleósidos de la transcriptasa reversa (INTR). Se desconoce la relevancia clínica de los aumentos transitorios del lactato en suero. También se han presentado reportes muy raros de retraso en el desarrollo, convulsiones y otras enfermedades neurológicas. Sin embargo, no se ha establecido una relación causal entre estos eventos y la exposición a INTR in utero o peri-parto. Estos hallazgos no afectan las recomendaciones actuales sobre el uso del tratamiento antirretroviral en mujeres embarazadas para evitar la transmisión vertical del VIH. Lactancia: Los expertos en salud recomiendan que siempre que sea posible, las mujeres infectadas con VIH no amamanten a sus hijos para evitar la transmisión del VIH. En escenarios en los que no es posible la alimentación con fórmula, deben seguirse los lineamientos oficiales locales sobre la lactancia y el tratamiento al considerar la lactancia durante el tratamiento con antirretrovirales. Aunque esto no ha sido confirmado en humanos, en base a los datos en animales, se espera que dolutegravir sea secretado en la leche humana. En un estudio, después de dosis orales repetidas de 150 mg de lamivudina 2 veces al día (administrada en combinación con 300 mg de zidovudina 2 veces al día) o 300 mg de lamivudina 2 veces al día, lamivudina se excretó en la leche materna en humanos (0.5 a 8.2 microgramos/ml) en concentraciones similares a las encontradas en suero. En otros estudios, después de dosis orales repetidas de 150 mg de lamivudina 2 veces al día (administradas combinadas con 300 mg de zidovudina o como Combivir o Tricivir), la relación leche materna: plasma materno varió entre 0.6 y 3.3. En un estudio, después de la administración oral repetida de 300 mg de abacavir 2 veces al día (administrado como Tricivir), la relación leche materna : plasma materno fue de 0.9. No se realizaron estudios farmacocinéticos con la administración oral 1 vez al día de abacavir. La mediana de las concentraciones séricas en lactantes de lamivudina varió entre 18 y 28 ng/ml, y estas no fueron detectables en uno de los estudios (sensibilidad del ensayo 7 ng/ml). La mayoría de los lactantes (8 de 9) no tenían niveles detectables de abacavir (sensibilidad del ensayo 16 ng/ml). No se midieron los niveles intracelulares de carbovir y lamivudina trifosfato (los metabolitos activos de abacavir y lamivudina) en lactantes alimentados con leche materna, por lo que se desconoce la relevancia de las concentraciones séricas de los compuestos principales medidos. Efectos sobre la capacidad para manejar y utilizar maquinaria: No se han realizado estudios para investigar el efecto de dolutegravir, abacavir o lamivudina sobre la capacidad para manejar o utilizar maquinaria. No se anticipa un efecto negativo sobre dichas actividades en base a la farmacología de estos productos medicinales. Debe tenerse en cuenta el estado clínico del paciente y el perfil de eventos adversos de Triumeq al considerar la capacidad del paciente para manejar o utilizar maquinaria. Carcinogénesis/mutagénesis: Dolutegravir no fue mutagénico ni clastogénico utilizando pruebas in vitro en bacterias y células cultivadas de mamíferos, y en un ensayo de micronúcleos de roedores in vivo. Dolutegravir no fue carcinogénico en los estudios a largo plazo en ratones y ratas. Ni abacavir ni lamivudina fueron mutagénicos en las pruebas bacterianas, pero al igual que varios análogos de nucleósidos, muestran actividad tanto en las pruebas de mamíferos in vitro como en el ensayo de linfoma en ratón. Esto es consistente con la actividad conocida de otros análogos de nucleósidos. Los resultados de una prueba de micronúcleos de ratas in vivo con abacavir y lamivudina combinados, fueron negativos. Los estudios de carcinogenicidad con abacavir administrado por vía oral en ratones y ratas, mostraron un aumento en la incidencia de tumores malignos y no malignos. Los tumores malignos ocurrieron en la glándula prepucial de machos y en la glándula clitorídea de hembras de ambas especies, y en el hígado, vejiga urinaria, ganglios linfáticos, y tejido subcutáneo de ratas hembra. La mayoría de estos tumores ocurrieron con la dosis más alta de abacavir de 330 mg/kg/día en ratones y de 600 mg/kg/día en ratas. Estos niveles de dosis fueron equivalentes a 21-28 veces la exposición sistémica esperada en humanos cuando abacavir se administró en combinación dolutegravir y lamivudina. La excepción fueron los tumores de la glándula prepucial en ratones, que ocurrieron con una dosis de 110 mg/kg. La exposición con esta dosis es de aproximadamente 5 veces la exposición sistémica en humanos. No existe una contraparte estructural de esta glándula en humanos. Al tiempo que se desconoce el potencial carcinogénico en humanos, estos datos sugieren que el riesgo carcinogénico en humanos es sobrepasado por el potencial beneficio clínico. Lamivudina no ha demostrado actividad genotóxica en los estudios in vivo. Los resultados de los estudios de carcinogenicidad a largo plazo en ratas y ratones, no mostraron potencial carcinogénico con exposiciones de aproximadamente 12 a 72 veces mayores que los niveles plasmáticos clínicos. Toxicología reproductiva: Fertilidad: Los estudios de fertilidad en ratas han demostrado que dolutegravir, abacavir y lamivudina no tienen un efecto sobre la fertilidad de machos ni hembras. Dolutegravir no afectó la fertilidad de machos ni hembras en ratas con dosis hasta de 1000 mg/kg/día, la dosis más alta evaluada (44 veces la exposición clínica en humanos con 50 mg cuando dolutegravir se administra en combinación con abacavir y lamivudina en base al AUC). Embarazo: En estudios de toxicidad reproductiva en animales, dolutegravir, abacavir y lamivudina demostraron cruzar la placenta. La administración oral de dolutegravir en ratas preńadas con dosis hasta de 1000 mg/kg día desde los días 6 a 17 de gestación, no desencadenó toxicidad materna, toxicidad del desarrollo ni teratogenicidad (50 veces la exposición clínica en humanos con 50 mg cuando dolutegravir se administra en combinación con abacavir y lamivudina, en base al AUC). La administración oral de dolutegravir en conejos preńados con dosis hasta de 1000 mg/kg diarios de los días 6 a 18 de gestación, no desencadenó toxicidad del desarrollo ni teratogenicidad (0.74 veces la exposición clínica en humanos con 50 mg cuando dolutegravir se administra combinado con abacavir y lamivudina, en base al AUC). En conejos, se observó toxicidad materna (disminución del consumo de alimentos, heces escasas/ausente, orina escasa, supresión de la ganancia de peso corporal) con 1000 mg/kg (0.74 veces la exposición clínica en humanos con 50 mg cuando dolutegravir se administra combinado con abacavir y lamivudina, en base al AUC). Abacavir demostró toxicidad para con el embrión en desarrollo y el feto solo en ratas con dosis tóxicas para la madre de 500 mg/kg/día y mayores. Esta dosis es de aproximadamente 28 veces la exposición terapéutica en humanos en base al AUC, para una dosis de 600 mg combinada con dolutegravir y lamivudina. Los hallazgos incluyeron edema fetal, variaciones y malformaciones, resorciones, disminución del peso corporal del feto y aumento de los mortinatos. La dosis en la que no se observaron efectos sobre el desarrollo pre o post-natal fue de 160 mg/kg/día. Esta dosis es equivalente a una exposición de aproximadamente 9 veces la de los humanos. No se observaron hallazgos similares en conejos. Lamivudina no fue teratogénica en los estudios en animales, pero se observaron indicaciones de un aumento de las muertes embriónicas tempranas en conejos con niveles de exposición comparables a los logrados en humanos. Sin embargo, no se observó evidencia de pérdida embrionaria en ratas con niveles de exposición de aproximadamente 32 veces la exposición clínica (en base a la Cmáx). Toxicología y/o farmacología en animales: Se ha evaluado el efecto del tratamiento diario prolongado con dosis altas de dolutegravir en estudios de toxicidad con dosis orales repetidas en ratas (hasta de 26 semanas) y en monos (hasta de 38 semanas). El efecto principal de dolutegravir fue intolerancia o irritación gastrointestinal en ratas y monos con dosis que producen exposiciones sistémicas de aproximadamente 38 y 1.5 veces la exposición clínica en humanos con 50 mg cuando dolutegravir se administra combinado con abacavir y lamivudina, en base al AUC, respectivamente. Debido a que la intolerancia gastrointestinal (IG) se considera secundaria a la administración local del fármaco, las métricas en mg/kg o mg/m2 son determinantes adecuados de la seguridad de esta toxicidad. La intolerancia GI en monos ocurrió con 30 veces la dosis equivalente en humanos en mg/kg (en base a un humano de 50 kg), y 11 veces la dosis equivalente en humanos en mg/m2 para una dosis clínica diaria total de 50 mg. Se observó degeneración miocárdica leve en el corazón de ratones y ratas después de la administración de abacavir durante 2 ańos. Las exposiciones sistémicas fueron equivalentes a 7 - 21 veces la exposición en humanos con 600 mg cuando abacavir se administra combinado con dolutegravir y lamivudina. No se ha determinado la relevancia clínica de este hallazgo.
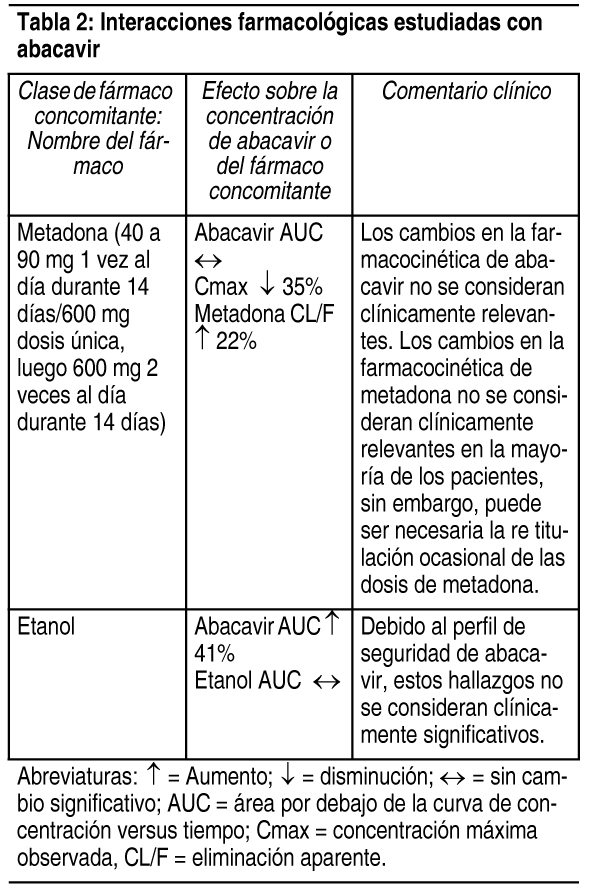
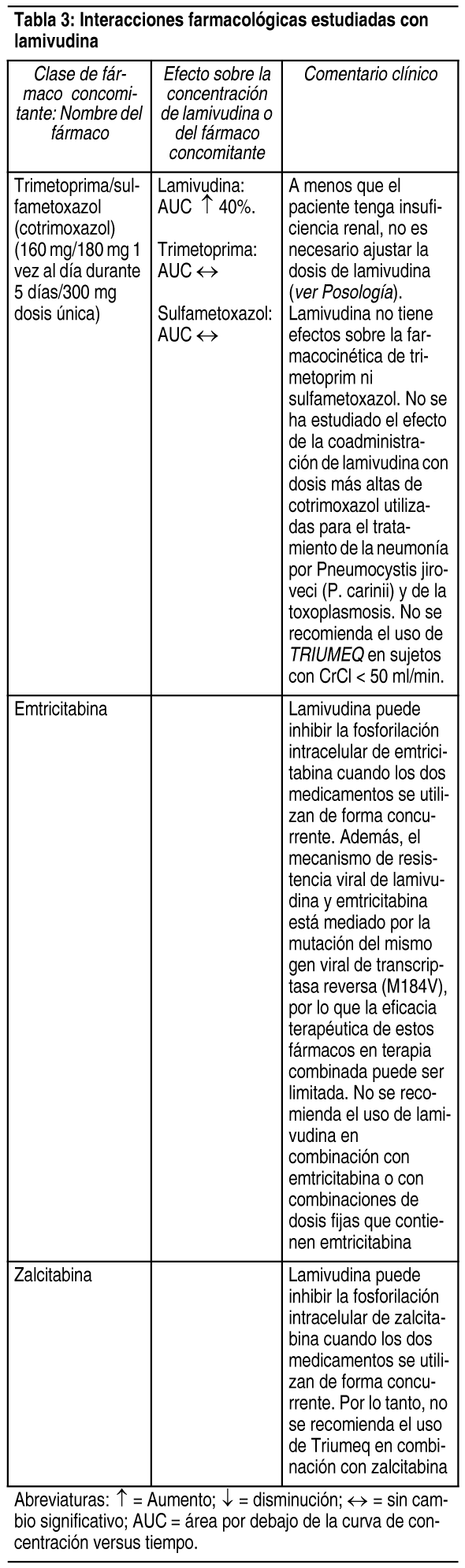
Interacciones Medicamentosas: Debido a que Triumeq contiene dolutegravir, abacavir y lamivudina, puede ocurrir con Triumeq cualquier interacción identificada con estos agentes de forma individual. Debido a las distintas vías de metabolismo y eliminación, no se esperan interacciones farmacológicas clínicamente significativas entre dolutegravir, abacavir y lamivudina. En una comparación cruzada entre estudios, las exposiciones a abacavir y lamivudina fueron similares al administrar Triumeq en comparación con Kivexa solo. Efecto de dolutegravir, abacavir y lamivudina sobre la farmacocinética de otros agentes: In vitro, dolutegravir demostró no inhibición directa, o débil (IC50>50 µM) de las enzimas del citocromo P450 (CYP)1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 CYP3A, uridina difosfato glucuronosil transferasa (UGT)1A1 o UGT2B7, o los transportadores Pgp, BCRP, BSEP, OATP1B1, OATP1B3, OCT1 MRP2, o MRP4. In vitro, dolutegravir no induce a CYP1A2, CYP2B6 o CYP3A4. In vivo, dolutegravir no tuvo un efecto sobre midazolam, un sustrato del CYP3A4. En base a estos datos, no se espera que dolutegravir afecte la farmacocinética de los fármacos que son sustratos de estas enzimas o transportadores. En los estudios de interacción farmacológica, dolutegravir no tuvo un efecto clínicamente relevante sobre la farmacocinética de los siguientes: tenofovir, ritonavir, metadona, efavirenz, lopinavir, atazanavir, darunavir, etravirina, fosamprenavir, rilpivirina, boceprevir, telaprevir, doclatasvir y anticonceptivos orales que contienen norgestimato y etinil estradiol. In vitro, dolutegravir inhibió al transportador renal de cationes orgánicos 2 (OCT2) (IC50 = 1.93 µM), al transportador de extrusión multifármacos y de toxinas (MATE) 1 (IC50 = 6.34 µM) y a MATE2-K (IC50 = 24.8 µM). Dada la exposición in vivo a dolutegravir, tiene un bajo potencial de afectar el transporte de sustratos de MATE2-K in vivo. In vivo, dolutegravir puede aumentar las concentraciones plasmáticas de los fármacos cuya excreción es dependiente de OCT2 o MATE1 (dofetilida, pilsicainida o metformina) (ver Tabla 1). In vitro, dolutegravir inhibió a los transportadores renales basolaterales: transportador de aniones orgánicos (OAT) 1 (IC50 = 2.12 µM) y OAT3 (IC50 = 1.97 µM). Sin embargo, dolutegravir no tuvo un efecto notable sobre la farmacocinética in vivo de los sustratos del OAT tenofovir y paraaminohipurato, por lo que existió poca propensión de ocasionar interacciones farmacológicas mediante la inhibición de los transportadores OAT. Abacavir y lamivudina no inhibieron ni indujeron a las enzimas CYP (como CYP 3A4, CYP 2C9 o CYP 2D6). Efecto de otros agentes sobre la farmacocinética de dolutegravir, abacavir y lamivudina: Dolutegravir se elimina principalmente mediante el metabolismo de UGT1A1. Dolutegravir también es un sustrato de UGT1A3, UGT1A9, CYP3A4, Pgp, y BCRP; por lo tanto, los fármacos que inducen estas enzimas o transportadores, en teoría pueden disminuir la concentración plasmática de dolutegravir y disminuir el efecto terapéutico de Triumeq. La coadministración de dolutegravir y otros fármacos que inhiben a UGT1A1, UGT1A3, UGT1A9, CYP3A4, y/o Pgp, puede aumentar la concentración plasmática de dolutegravir (ver Tabla 1). In vitro, dolutegravir no es un sustrato del polipéptido transportador de aniones orgánicos humanos (OATP) 1B1, OATP1B3, u OCT1; por lo tanto, no se espera que los fármacos que únicamente modulen estos transportadores afecten la concentración plasmática de dolutegravir. Efavirenz, etravirina, nevirapina, rifampicina, carbamazepina y tipranavir en combinación con ritonavir, cada uno, disminuyeron las concentraciones plasmáticas de dolutegravir de forma significativa, y requirieron un ajuste de la dosis de dolutegravir a 50 mg 2 veces al día. El efecto de etravirina fue mitigado por la coadministración de los inhibidores de CYP3A4 lopinavir/ritonavir, darunavir/ritonavir, y se espera que sea mitigado por atazanavir/ritonavir. Por lo tanto, no es necesario ajustar la dosis de dolutegravir al coadministrarlo con etravirina y lopinavir/ritonavir, darunavir/ritonavir, o atazanavir/ritonavir. Otro inductor, fosamprenavir en combinación con ritonavir, disminuyó las concentraciones plasmáticas de dolutegravir, pero no requiere un ajuste de la dosis de dolutegravir. Un estudio de interacción farmacológica con el inhibidor de UGT1A1, atazanavir, no resultó en un aumento clínicamente significativo de las concentraciones plasmáticas de dolutegravir. Tenofovir, lopinavir/ritonavir, darunavir/ritonavir, rilpivirina, boceprevir, telaprevir, prednisona, rifabutina, daclatasvir y omeprazol, no tuvieron un efecto o este fue mínimo sobre la farmacocinética de dolutegravir, por lo que no es necesario ajustar la dosis de dolutegravir cuando se co administra con estos fármacos. La probabilidad de interacciones metabólicas con abacavir y lamivudina es baja. Abacavir y lamivudina no se metabolizaron de forma significativa mediante las enzimas CYP. Las principales vías del metabolismo de abacavir en humanos son por el alcohol deshidrogenasa y por la glucuronidación para producir 5'-ácido carboxílico y 5'-glucurónido, los cuales representan cerca del 66% de la dosis administrada. Estos metabolitos se excretan en la orina. La probabilidad de interacciones metabólicas con lamivudina es baja, debido al metabolismo limitado y a la unión a proteínas plasmáticas, así como a la eliminación casi completa por vía renal. Lamivudina se elimina principalmente mediante secreción activa de cationes orgánicos. Debe considerarse la posibilidad de interacciones con otros medicamentos administrados de forma concurrente, particularmente cuando la principal vía de eliminación es renal. Triumeq es 1 comprimido con dosis fijas y no deberá prescribirse en pacientes que requieren ajustes de la dosis debido a medicamentos concomitantes que ocasionan interacciones. Las preparaciones por separado de dolutegravir, abacavir y lamivudina deben administrarse en los casos en los que se requiere ajustar la dosis. En estos casos, el médico debe ver la información individual del producto para estos medicamentos. Se presentan las interacciones farmacológicas seleccionadas en las Tablas 1, 2 y 3. Las recomendaciones están basadas tanto en estudios de interacciones farmacológicas o en las interacciones predichas debido a la magnitud esperada de la interacción y el potencial de eventos adversos serios o la pérdida de eficacia.
Sobredosificación:Síntomas y signos: Actualmente existe experiencia limitada con la sobredosis de dolutegravir. La experiencia limitada con dosis únicas más altas (hasta de 250 mg en sujetos sanos), no reveló signos o síntomas específicos además de los detallados como reacciones adversas. No se han identificado signos o síntomas específicos después de la sobredosis aguda con abacavir o lamivudina, además de los detallados como reacciones adversas. Tratamiento: El manejo posterior debe ser según esté indicado clínicamente o según las recomendaciones del centro nacional de toxicología, donde exista. Si ocurre una sobredosis, el paciente debe recibir tratamiento de apoyo con monitoreo adecuado según sea necesario. Debido a que lamivudina es dializable, puede utilizarse hemodiálisis continua para el tratamiento de la sobredosis, aunque esto no ha sido estudiado. Se desconoce si abacavir puede eliminarse mediante diálisis peritoneal o hemodiálisis. Debido a que dolutegravir se une en alto grado a las proteínas plasmáticas, es poco probable que se elimine de forma significativa mediante diálisis.
Incompatibilidades: No se han identificado incompatibilidades.
Conservación:Vida de anaquel: 24 meses. Precauciones especiales para el almacenamiento: No almacene a más de 30°C. Almacene en el empaque original para protegerlo de la humedad. Mantenga el frasco bien cerrado. No retire el desecante.
Observaciones: Triumeq, Tvicay, Ziagen, Epivir, Kivexa, Tricivir, Combivir son marcas registradas del grupo de compańías ViiV Healthcare.
Presentaciones:Naturaleza y contenidos del envase: Los comprimidos de Triumeq están disponibles en frascos de polietileno blanco de alta densidad (HDPE) que contienen un desecante.
 Laboratorio
Laboratorio