Composición:Frasco-ampolla: Cada frasco-ampolla contiene: 30 MU (0.300 mg) de Filgrastim. Jeringas prellenadas: Cada jeringa prellenada contiene: 30 MU (0.300 mg) de Filgrastim. Excipientes: Acido Acético Glacial, Hidróxido de Sodio, Sorbitol, Polisorbato 80, Agua para Inyectables.
Acción Terapéutica: Factores estimulantes de colonia.
Indicaciones: Neupogen está indicado para reducir la duración de la neutropenia y la incidencia de neutropenia febril en pacientes tratados con quimioterapia citotóxica convencional con enfermedades malignas (con la excepción de leucemia mieloide crónica y síndromes mielodisplásicos) y para la reducción de la duración de la neutropenia en los pacientes sometidos a tratamiento mieloablativo seguido de trasplante de médula ósea y que se considere presenten un mayor riesgo de experimentar neutropenia grave prolongada. La eficacia y la seguridad de Neupogen son similares en adultos y en nińos que están recibiendo quimioterapia citotóxica. Neupogen está indicado para la movilización de células progenitoras hematopoyéticas en sangre periférica (PBPC's). En pacientes, tanto nińos como adultos, con neutropenia congénita grave, cíclica o idiopática con una cuenta absoluta de neutrófilos (CAN) de ≤0.5 x 109/l, y antecedentes de infecciones graves o recurrentes, la administración prolongada de Neupogen está indicada para aumentar la cuenta de neutrófilos y reducir la incidencia y duración de episodios infecciosos. Neupogen está indicado para el tratamiento de la neutropenia persistente (CAN igual o inferior a 1.0 x 109/l) en pacientes con infección avanzada por el VIH, para reducir el riesgo de desarrollar infecciones bacterianas cuando otras opciones para tratar la neutropenia no sean adecuadas.
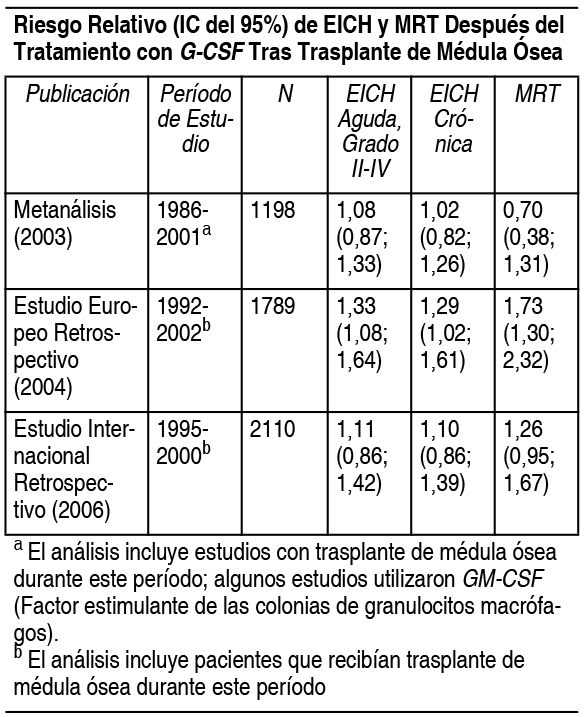
Propiedades:Acción (Farmacología y/o Terapéutica) los modos de acción del medicamento en el hombre: Farmacología: El factor estimulante de las colonias de granulocitos humano (G-CSF) es una glucoproteína que regula la producción y liberación de los neutrófilos funcionales de la médula ósea. Neupogen contiene r-metHuG-CSF (filgrastim) que aumenta considerablemente la cuenta de neutrófilos en sangre periférica a las 24 horas, con un aumento mínimo del número de monocitos. Filgrastim también puede inducir un leve aumento de los eosinófilos y basófilos circulantes con relación a los valores iniciales en algunos pacientes con neutropenia crónica grave (NCG); algunos de estos pacientes muestran eosinofilia o basofilia ya antes del tratamiento. El incremento de los neutrófilos depende de la dosis, cuando se aplica la posología recomendada. Los neutrófilos producidos en respuesta a filgrastim muestran una función normal o superior a la habitual, de acuerdo con las pruebas de la función quimiotáctica y fagocitaria. Después de interrumpir el tratamiento con filgrastim, la cuenta de neutrófilos circulantes se reduce un 50% al cabo de 1-2 días y se normaliza en un plazo de 1 a 7 días. El empleo de filgrastim en pacientes sometidos a quimioterapia citotóxica lleva a reduciones significativas en la incidencia, gravedad y duración de la neutropenia y de la neutropenia febril. El tratamiento con filgrastim reduce significativamente la duración de la neutropenia febril, el uso de antibióticos y la hospitalización, tras la quimioterapia de inducción en la leucemia mieloide aguda seguida de trasplante de médula ósea. La incidencia de fiebre e infecciones documentadas no disminuyó en estas condiciones clínicas. No se redujo la duración de la fiebre en los pacientes sometidos a terapia mieloablativa seguida de trasplante de médula ósea. La administración de filgrastim, ya sea administrado solo o tras la quimioterapia, moviliza las células progenitoras hematopoyéticas hacia la sangre periférica. De este modo, resulta posible obtener estas células progenitoras en sangre periférica (PBPC's) autóloga e infundirlas en el propio paciente tras la quimioterapia citotóxica intensiva, ya sea en lugar del trasplante de médula ósea o además de éste. Dado que la perfusión de células progenitoras hematopoyéticas (PBPC) acelera la recuperación hematopoyética, reduce la duración del riesgo de complicaciones hemorrágicas y la necesidad de transfusiones plaquetarias. Los pacientes que recibieron células progenitoras hematopoyéticas de sangre periférica (PBPC's) alógenicas movilizadas con Neupogen experimentaron una recuperación hematopoyética significativamente más rápida, lo cual se tradujo en un descenso importante del tiempo transcurrido hasta la recuperación de las plaquetas sin apoyo en comparación con los pacientes sometidos a trasplante de médula ósea. Un estudio europeo, retrospectivo, que evaluó el uso de G-CSF tras la realización de trasplante alogénico de médula ósea en pacientes con leucemia aguda sugirió un aumento en el riesgo de enfermedad de injerto contra huésped (EICH), mortalidad relacionada con el tratamiento (MRT) y mortalidad cuando se administró G-CSF. En otro estudio retrospectivo internacional en pacientes con leucemia mieloide aguda y crónica, no se observó efecto sobre el riesgo de enfermedad de injerto contra el huésped, MRT, ni mortalidad. Un metanálisis de estudios de trasplantes alogénicos, incluyendo los resultados de 9 ensayos prospectivos de asignación aleatoria, 8 estudios retrospectivos y 1 estudio caso-control, no detectaron un efecto en el riesgo de enfermedad aguda de injerto contra el huésped, enfermedad crónica de injerto contra el huésped o mortalidad temprana relacionada con el tratamiento.
Utilización de filgrastim para la movilización de células progenitoras hematopoyéticas (PBPC's) en donantes sanos previo al trasplante alogénico de células progenitoras hematopoyéticas: En donantes sanos, la administración subcutánea de 10 mcg/kg/día durante 4 a 5 días consecutivos permite obtener ≥4 x 106 células CD34+/kg de peso corporal del receptor en la mayoría de los donantes después de 2 leucocitaféresis. El empleo de filgrastim en pacientes, tanto nińos como adultos, con NCG (neutropenia congénita grave, neutropenia cíclica y neutropenia idiopática) induce un aumento sostenido en la cuenta absoluta de neutrófilos en sangre periférica y una reducción en el número de infecciones y procesos relacionados. La administración de filgrastim a pacientes con infección por VIH mantiene la cuenta de neutrófilos en los niveles normales permitiendo la administración de la medicación antiviral y/u otras medicinas mielosupresoras. No hay evidencia de que los pacientes con infección por VIH tratados con filgrastim presenten un aumento en la replicación del VIH. Como otros factores de crecimiento hematopoyéticos, el G-CSF han demostrado in vitro tener propiedades estimulantes sobre las células endoteliales humanas. Farmacocinética: El aclaramiento o eliminación de filgrastim sigue una farmacocinética de primer orden, tras su administración subcutánea e intravenosa. La vida media de eliminación de filgrastim en suero es de aproximadamente 3.5 horas con una tasa de aclaramiento aproximada de 0.6 mlmin/kg. La perfusión continua de Neupogen a lo largo de períodos de hasta 28 días en pacientes que se recuperan del trasplante de médula ósea autólogo no se asocia a acumulación farmacológica y las semividas de eliminación son comparables. Existe una correlación lineal positiva entre la dosis y la concentración sérica de filgrastim tanto si se administra por vía intravenosa como subcutánea. Las concentraciones séricas se mantienen por encima de 10 ng/ml durante 8 a 16 horas después de la administración subcutánea de las dosis recomendadas. El volumen de distribución en sangre es de aproximadamente 150 ml/kg.
Posología:Dosis y administración: La terapia con Neupogen solo debe administrarse en colaboración con un centro oncológico con experiencia en el tratamiento con G-CSF y hematología, y que tenga las instalaciones diagnósticas necesarias. Los procedimientos de movilización y aféresis deben realizarse en colaboración con un centro hemato-oncológico con experiencia aceptable en este campo y donde el monitoreo del progenitor hematopoyético pueda realizarse correctamente. Quimioterapia citotóxica: Posología: La dosis recomendada de Neupogen es de 0.5 MU (5 mcg)/kg/día. La primera dosis de Neupogen deberá administrarse a partir de las 24 horas siguientes de finalizada la quimioterapia citotóxica. En los estudios clínicos aleatorizados se utilizó una dosis subcutánea de 230 mcg/m2/día (4.0 a 8.4 mcg/kg/día). El tratamiento diario con Neupogen debe continuarse hasta que se haya superado el punto mínimo previsto para la cuenta de neutrófilos y éste haya vuelto a valores normales. Tras la quimioterapia convencional en tumores sólidos, linfomas y leucemias linfocíticas, se requiere un tratamiento de hasta 14 días para alcanzar este objetivo. Después del tratamiento de inducción y consolidación de la leucemia mieloide aguda, la duración del tratamiento puede ser considerablemente mayor (hasta de 38 días) dependiendo del tipo, la dosis y el esquema de quimioterapia aplicado. En los pacientes sometidos a quimioterapia citotóxica suele observarse un aumento transitorio en la cuenta de neutrófilos que ocurre típicamente 1-2 días después de iniciar el tratamiento de Neupogen. Sin embargo, para una respuesta terapéutica sostenida, no se debe suspender el tratamiento con Neupogen hasta que haya superado el punto mínimo previsto para que la cifra de neutrófilos regrese a valores normales. No se recomienda, por tanto, la interrupción prematura del tratamiento con Neupogen antes de haber pasado el punto mínimo previsto para la cifra de neutrófilos. Método de administración: Neupogen puede administrarse 1 vez al día en forma de inyección subcutánea o diluido en solución glucosada al 5% como perfusión intravenosa administrada en 30 minutos. La vía subcutánea es preferida en la mayoría de los casos. De acuerdo con un estudio de administración de dosis únicas, la vía intravenosa puede acortar la duración del efecto. Se desconoce cuál podría ser la relevancia clínica de este dato para la administración del fármaco en dosis múltiples. La elección de la vía de administración más adecuada dependerá de las circunstancias clínicas de cada paciente. No se recomienda en ningún caso diluir a concentraciones finales inferiores a 0.2 MU (2 mcg) por ml. La solución debe inspeccionarse visualmente antes de usarla. Solamente deben utilizarse soluciones transparentes sin partículas. Para pacientes tratados con filgrastim diluido a concentraciones inferiores a 1.5 MU (15 mcg) por ml, debe ańadirse albúmina sérica humana para una concentración final de 2 mg/ml. Ejemplo: Si el volumen de inyección final es de 20 ml, las dosis totales de filgrastim inferiores a 30 MU (300 mcg), deben administrarse ańadiendo 0.2 ml de solución de albúmina humana al 20% (F. Eur.). Neupogen no contiene preservadores. En vista de un posible riesgo de contaminación microbiana, las jeringas prellenadas de Neupogen son para un solo uso. Neupogen, diluido en glucosa al 5%, es compatible con el vidrio y diversos plásticos como PVC, poliolefina (copolímero de polipropileno y polietileno) y polipropileno. La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él, se realizará de acuerdo con la normativa local. Pacientes tratados con terapia mieloablativa seguida por trasplante de médula ósea: Posología: La dosis inicial recomendada de Neupogen es de 1.0 MU (10 mcg)/kg/día. La primera dosis de Neupogen debe aplicarse al menos 24 horas después de la quimioterapia citotóxica y al menos 24 horas después de la perfusión de la médula ósea. Una vez superado el punto mínimo esperado en la cuenta de neutrófilos, la dosis diaria de Neupogen debe ajustarse de acuerdo con la respuesta de los neutrófilos de la siguiente forma: Cuenta Absoluta de Neutrófilos (CAN): Ajuste de la Dosis de Neupogen. >1.0 x 109/l durante 3 días consecutivos: Disminuir a 0.5 MU (5 mcg)/kg/día. Si la CAN permanece >1.0 x 109/l durante 3 días consecutivos más: Suspender Neupogen. Si el CAN desciende a <1.0 x 109/l durante el tratamiento: Aumentar la dosis según los pasos arriba indicados. CAN = cuenta absoluta de neutrófilos. Método de administración: Neupogen puede ser administrado como perfusión intravenosa en 30 minutos o 24 horas, o bien, administrado por perfusión subcutánea continua por 24 horas. Neupogen debe ser diluido en 20 ml de solución de glucosa al 5%. Para la movilización de células progenitoras hematopoyéticas en sangre periférica (PBPC's) en pacientes sometidos a terapia mielosupresora o mieloablativa seguida de trasplante autólogo de PBPC. Posología: La dosis recomendada de Neupogen para la movilización de PBPC, cuando se utiliza solo, es de 1.0 MU (10 mcg)/kg/día durante 5 a 7 días consecutivos. Ritmo en leucocitaféresis: generalmente suele bastar con 1 ó 2 leucocitaféresis en los días 5 y 6. En otras circunstancias, puede ser necesaria leucocitaféresis adicional. La administración de Neupogen debe mantenerse hasta la última leucocitaféresis. La dosis recomendada de Neupogen, para movilizar PBPC tras una quimioterapia mielosupresora, es de 0.5 MU (5 mcg)/kg/día, desde el primer día tras concluir la quimioterapia hasta que se haya superado el punto mínimo previsto de la cuenta de neutrófilos y este haya vuelto a los valores normales. Se debe realizar la leucocitaféresis en el período comprendido en que la CAN aumenta desde ≤0.5 x 109/l a >5.0 x 109/l. En aquellos pacientes que no hayan sido sometidos a quimioterapia intensiva, una leucocitaféresis suele ser suficiente. En otras circunstancias, se recomiendan leucocitaféresis adicionales. Método de administración: Neupogen para la movilización de PBPC, cuando se utiliza solo: Neupogen puede administrarse como perfusión subcutánea continúa por 24 horas o inyección subcutánea. Para las perfusiones, Neupogen debe ser diluido en 20 ml de solución glucosa al 5%. Neupogen para la movilización de PBPC posterior a quimioterapia mielosupresora: Neupogen debe ser administrado mediante inyección subcutánea. Para la movilización de células hematopoyéticas en donantes sanos de forma previa al trasplante alogénico de células progenitoras hematopoyéticas periféricas (PBPC): Posología: Para la movilización de PBPC en donantes sanos, Neupogen debe administrarse a dosis de 1.0 MU (10 mcg)/kg/día durante 4 ó 5 días consecutivos. La leucocitaféresis debe iniciarse el día 5 y continuarse el día 6 si fuera necesario para obtener 4 x 106 células CD34+/kg de peso corporal del receptor. Método de administración: Neupogen debe ser administrado por inyección subcutánea. En pacientes con NCG: Posología: Neutropenia congénita: la dosis inicial recomendada es de 1.2 MU (12 mcg)/kg/día, que se administra como dosis única o en dosis divididas. Neutropenia idiopática o cíclica: la dosis inicial recomendada es de 0.5 MU (5 mcg)/kg/día, que se administra en dosis única o repartida en dosis divididas. Ajuste de la dosis: Neupogen debe administrarse diariamente por inyección subcutánea hasta que la cuenta de neutrófilos se estabilice por encima de 1.5 x 109/l. Una vez alcanzada la respuesta debe determinarse la dosis mínima efectiva para mantener esta cifra. La administración diaria y prolongada es requerida para mantener una cuenta de neutrófilos adecuada. Al cabo de 1 a 2 semanas de tratamiento, la dosis inicial puede reducirse a la mitad o aumentarse al doble, dependiendo de la respuesta del paciente. A partir de entonces, la dosis se puede ajustar en forma individual cada 1 a 2 semanas con el fin de mantener la cifra de neutrófilos entre 1.5 x 109/l y 10 x 109/l. En pacientes con infecciones graves se puede considerar un aumento más rápido de la dosis. En los ensayos clínicos, el 97% de los pacientes que respondieron al tratamiento presentaron una respuesta completa a dosis ≤24 mcg/kg/día. En pacientes con neutropenia crónica grave (NCG), no se ha establecido la seguridad a largo plazo de la administración de Neupogen con dosis superiores a 24 mcg/kg/día. Método de administración: En neutropenia congénita, idiopática o cíclica, Neupogen debe ser administrado por inyección subcutánea. En pacientes con infección por VIH: Posología: Para corregir la neutropenia: La dosis inicial recomendada de Neupogen es 0.1 MU (1 mcg)/kg/día ajustando hasta un máximo de 0.4 MU (4 mcg)/kg/día hasta alcanzar y mantener una cifra normal de neutrófilos (CAN > 2.0 x 109/l). En los ensayos clínicos, > 90% de los pacientes respondieron a estas dosis, recuperándose de la neutropenia en un tiempo mediano de 2 días. En un pequeńo número de pacientes (<10%) se necesitaron dosis de hasta 1.0 MU (10 mcg)/kg/día para corregir la neutropenia. Para mantener la cifra de neutrófilos dentro de la normalidad: Una vez corregida la neutropenia, debe determinarse la dosis mínima eficaz necesaria para mantener una cifra normal de neutrófilos. Se recomienda ajustar la dosis inicial a 30 MU (300 mcg)/día a días alternos. En ocasiones puede ser necesario seguir ajustando la dosis de acuerdo con la CAN del paciente, para mantener el número de neutrófilos > 2.0 x 109/l. En los ensayos clínicos, se requirió la administración de 30 MU (300 mcg)/día de 1 a 7 días por semana para mantener la CAN > 2.0 x 109/l, siendo la mediana en la frecuencia de administración de 3 días a la semana. Puede ser necesaria una administración a largo plazo para mantener la CAN > 2.0 x 109/l. Método de administración: Para la recuperación de la neutropenia o mantener una cuenta normal de neutrófilos: Neupogen debe ser administrado por inyección subcutánea. Personas de edad avanzada: Los estudios clínicos con Neupogen han incluido a un número pequeńo de pacientes ancianos pero estudios especiales no se han realizado en este grupo, por lo tanto, no pueden hacerse recomendaciones específicas de posología. Pacientes con insuficiencia renal: Los estudios con Neupogen en pacientes con insuficiencia renal o función hepática grave demuestran un perfil farmacocinético y farmadinámico similar al observado en individuos normales. El ajuste de dosis no es requerido en estas circunstancias. Uso pediátrico en la NCG y cáncer: 65% de los pacientes estudiados en el programa de ensayos clínicos sobre NCG fueron menores de 18 ańos. La eficacia del tratamiento fue evidente en este grupo de edad, que incluyó a una mayoría de pacientes con neutropenia congénita. No se observó ninguna diferencia en el perfil de seguridad en los pacientes pediátricos en tratamiento para NCG. Los datos procedentes de ensayos clínicos en pacientes pediátricos indican que la seguridad y eficacia de Neupogen son similares en adultos y nińos tratados con quimioterapia citotóxica. Las dosis recomendadas en pacientes pediátricos tratados con quimioterapia citotóxica mielosupresora son las mismas que en adultos.
Modo de Empleo:Instrucciones especiales de uso, manipulación y eliminación: Evítense las sacudidas violentas. La solución debe inspeccionarse visualmente antes de usarla. Sólo se utilizará si está límpida y sin partículas. Los viales y las jeringas precargadas de filgrastim son de un solo uso. Istrucciones para la dilución: En caso necesario, puede diluirse el filgrastim en solución glucosada al 5%. No se recomienda diluirlo en ningún caso hasta concentraciones finales inferiores a 5 µg/ml. Para los pacientes tratados con Neupogen diluido en concentraciones inferiores a 1.5 MU/ml (15 µg/ml) debe ańadirse albúmina sérica humana (ASH, F. Eur.) hasta alcanzar una concentración final de 2 mg/ml. Ejemplo: Si el volumen de inyección final es de 20 ml y la dosis total de Neupogen inferior a 30 MU (300 µg), deben administrarse 0.2 ml de una solución de albúmina humana al 20%. Las soluciones diluidas de Neupogen no deben prepararse más de 24 horas antes de la administración y deben conservarse refrigeradas a 2-8 şC. Eliminación de medicamentos no utilizados/caducados: La emisión de productos farmacéuticos al medio ambiente debe reducirse a un mínimo. Los medicamentos no deben eliminarse a través de las aguas residuales, y su eliminación con los residuos domésticos también debe evitarse. Utilice los sistemas de recogida establecidos si los hay en su localidad.
Efectos Colaterales:A. Resumen del perfil de seguridad: En los ensayos clínicos en pacientes con cáncer, la reacción adversa más frecuente fue el dolor músculo esquelético, que ocurrió de forma leve o moderada en 10% de los pacientes y de forma grave en 3%. También se ha notificado Enfermedad de Injerto Contra Huésped (EICH) (ver sección c más adelante). En la movilización de PBPC en donantes sanos, la reacción adversa notificada con mayor frecuencia fue el dolor músculo-esquelético. En donantes se ha observado leucocitosis, trombocitopenia tras la administración de filgrastim y leucocitaféresis. También se han notificado casos de esplenomegalia y ruptura esplénica. Algunos casos de ruptura esplénica fueron fatales. En pacientes con NCG las reacciones adversas más frecuentemente relacionadas con Neupogen fueron dolor óseo, dolor músculo-esquelético general y esplenomegalia. Pacientes con neutropenia congénita que reciben tratamiento con Neupogen han desarrollado SMD o leucemia (ver Precauciones). Se ha notificado como una reacción poco frecuente (≥1/1.000 a <1/100) el síndrome de fuga capilar, que puede poner en peligro la vida si se retrasa el tratamiento, en pacientes con cáncer sometidos a quimioterapia y en donadores sanos sometidos a movilización de las células progenitoras de sangre periférica, tras la administración de factores estimulantes de colonias de granulocitos (ver Precauciones y la subsección C de Efectos colaterales). En los ensayos clínicos en pacientes con VIH, las únicas reacciones adversas que se consideraron consistentemente relacionadas con la administración de Neupogen fueron el dolor músculo-esquelético, dolor óseo y mialgia. a. Resumen tabulado de reacciones adversas: Los datos incluidos en las siguientes tablas describen reacciones adversas notificadas durante ensayos clínicos y por notificación espontánea. Las reacciones adversas se enumeran en orden decreciente de gravedad dentro de cada intervalo de frecuencia. Se presentan los datos separados para los pacientes con cáncer, movilización de PBPC en donantes sanos, pacientes con NCG y pacientes con VIH, reflejando los diferentes perfiles de reacciones adversas en estas poblaciones. Pacientes con cáncer: Reacciones adversas: Muy frecuentes (≥1/10); Frecuentes (≥1/100 a <1/10); Poco frecuentes (≥1/1.000 a <1/100); Raras (≥1/10.000 a <1/1.000); Muy raras (<1/10.000). Trastornos de la sangre y del sistema linfático: Poco frecuentes: Ruptura esplénicaa; esplenomegaliaa,e, crisis de células falciformesa. Trastornos del sistema inmunológico: Frecuentes: Hipersensibilidad al medicamentoa. Poco frecuentes: Enfermedad del injerto contra huéspedb. Trastornos del metabolismo y de la nutrición: Ácido úrico elevado en sangre; deshidrogenasa láctica elevada en sangre; disminución del apetitoa. Poco frecuentes: Pseudogotaa. Trastornos del sistema nervioso: Muy frecuentes: Cefaleaa. Trastornos vasculares: Frecuentes: Hipotensión. Poco frecuentes: Enfermedad venoclusivad; alteración en el volumen de los fluidos; síndrome de fuga capilara. Trastornos respiratorios, torácicos y mediastínicos: Muy frecuentes: Dolor orofaríngeoa; tosa; disnea. Frecuentes: Hemoptisise. Poco frecuentes: Síndrome de distrés respiratorio agudoa; insuficiencia respiratoriaa; edema pulmonara; enfermedad pulmonar intersticiala; infiltración pulmonara; hemorragia pulmonar. Trastornos gastrointestinales: Muy frecuentes: Diarreaa; vómitosa; estreńimientoa; náuseaa. Trastornos hepatobiliares: Muy frecuentes: Gamma-glutamil transferasa elevada; fosfatasa alcalina elevada en sangre. Trastornos de la piel y del tejido subcutáneo: Muy frecuentes: Erupcióna, alopeciaa. Poco frecuentes: Síndrome de Sweet; vasculitis cutáneaa. Trastornos musculoesqueléticos y del tejido conjuntivo: Muy frecuentes: Dolor musculoesqueléticoc. Poco frecuentes: Exacerbación de artritis reumatoide. Trastornos renales y urinarios:Frecuentes: Disuria. Poco frecuentes: Anormalidad urinaria; glomerulonefritis. Trastornos generales y alteraciones en el sitio de administración: Asteniaa; fatigaa; inflamación de la mucosaa; Dolora. Frecuentes: dolor torácicoa. aVer sección c. b Se han notificado casos de EICH y muertes en pacientes tras el trasplante alogénico de médula ósea (ver sección c). c Incluye dolor óseo, dolor de espalda, artralgia, mialgia, dolor en extremidades, dolor músculo-esquelético, dolor torácico músculo-esquelético, cervicalgia. d Casos observados en pacientes sometidos a trasplante de médula ósea o movilización de PBPC en la experiencia postcomercialización. e Casos observados en el entorno del ensayo clínico. Movilización de PBPC en donantes sanos: Reacciones adversas: Muy frecuentes (≥1/10); Frecuentes (≥1/100 a <1/10); Poco frecuentes (≥1/1.000 a <1/100); Raras (≥1/10.000 a <1/1.000); Muy raras (<1/10.000). Trastornos de la sangre y del sistema linfático: Muy frecuentes: Trombocitopeniaa; leucocitosisa. Frecuentes: Esplenomegaliaa. Poco frecuentes: Ruptura esplénicaa; crisis de células falciformesa. Trastornos del sistema inmunológico: Poco frecuentes: Reacción anafiláctica. Trastornos del metabolismo y de la nutrición: Frecuentes: Deshidrogenasa láctica elevada en sangre. Poco frecuentes: Hiperuricemia (ácido úrico elevado en sangre). Trastornos del sistema nervioso: Muy frecuentes: Cefalea. Trastornos vasculares: Poco frecuentes: Síndrome de fuga capilara. Trastornos respiratorios, torácicos y mediastínicos: Frecuentes: Disnea. Poco frecuentes: Hemorragia pulmonar; hemoptisis; infiltración pulmonar; hipoxia. Trastornos hepatobiliares: Frecuentes: Fosfatasa alcalina elevada en sangre. Poco frecuentes: Aspartato aminotransferasa elevada. Trastornos músculo-esqueléticos y del tejido conjuntivo: Muy frecuentes: Dolor músculo-esqueléticob. Poco frecuentes: Exacerbación de artritis reumatoide. Trastornos renales y urinarios: Poco frecuentes: Glomerulonefritis. a Ver sección c. b incluye dolor óseo, dolor de espalda, artralgia, mialgia, dolor en una extremidad, dolor músculo-esquelético, dolor torácico músculo-esquelético, cervicalgia. Pacientes con NCG: Reacciones adversas: Muy frecuentes (≥ 1/10); Frecuentes (≥1/100 a <1/10); Poco frecuentes (≥1/1.000 a < 1/100); Raras (≥1/10.000 a <1/1.000); Muy raras (<1/10.000). Trastornos de la sangre y del sistema linfático: Esplenomegaliaa. Anemia. Frecuentes: Ruptura esplénicaa. Trombocitopeniaa. Poco frecuentes: Crisis de células falciformesa. Trastornos del metabolismo y de la nutrición: Muy frecuentes: Hiperuricemia; glucosa disminuida en sangre; deshidrogenasa láctica elevada en sangre. Trastornos del sistema nervioso: Muy frecuentes: Cefalea. Trastornos respiratorios, torácicos y mediastínicos: Muy frecuentes: Epistaxis. Trastornos gastrointestinales: Muy frecuentes: Diarrea. Trastornos hepatobiliares: Hepatomegalia; Fosfatasa alcalina elevada en sangre. Trastornos de la piel y del tejido subcutáneo: Muy frecuentes: Erupción. Frecuentes: Vasculitis cutánea. Alopecia. Trastornos músculo-esqueléticos y del tejido conjuntivo: Muy frecuentes: Dolor músculo-esqueléticob, artralgia. Frecuentes: Osteoporosis. Trastornos renales y urinarios: Hematuria; glomerulonefritis. Poco frecuentes: Proteinuria. Trastornos generales y alteraciones en el sitio de administración: Frecuentes: Dolor en el sitio de inyección. a Ver sección c. b incluye dolor óseo, dolor de espalda, artralgia, mialgia, dolor en una extremidad, dolor músculo-esquelético, dolor torácico músculo-esquelético, cervicalgia. Pacientes con VIH: Reacciones adversas: Muy frecuentes (≥1/10); Frecuentes (≥1/100 a <1/10); Poco frecuentes (≥1/1.000 a <1/100); Raras (≥1/10.000 a <1/1.000); Muy raras (<1/10.000); Se desconoce (no pueden ser estimadas con los datos disponibles). Trastornos de la sangre y del sistema linfático: Frecuentes: Esplenomegaliaa. Poco frecuentes: Crisis de células falciformesa. Trastornos músculo-esqueléticos y del tejido conjuntivo: Muy frecuentes: Dolor músculo-esqueléticob. Trastornos renales y urinarios: Se desconoce: Glomerulonefritis. a Ver sección c. b incluye dolor óseo, dolor de espalda, artralgia, mialgia, dolor en una extremidad, dolor músculo-esquelético, dolor torácico músculo-esquelético, cervicalgia. a. Descripción de las reacciones adversas seleccionadas: Se han notificado casos de enfermedad de injerto contra huésped (EICH) y muertes en pacientes que recibieron G-CSF tras el trasplante alogénico de médula ósea (ver Precauciones y Farmacología). Se han notificado casos de síndrome de fuga capilar en la experiencia postcomercialización con el uso de G-CSF. Estos casos ocurrieron generalmente en pacientes con enfermedades malignas avanzadas, sepsis, tratados con múltiples medicamentos quimioterapéuticos o sometidos a aféresis (ver Precauciones). Pacientes con cáncer: En los ensayos clínicos aleatorizados y controlados con placebo, Neupogen no aumentó la incidencia de reacciones adversas asociadas a la quimioterapia citotóxica. En estos estudios clínicos, los efectos adversos que ocurrieron con la misma frecuencia en los pacientes tratados con Neupogen/quimioterapia y placebo/quimioterapia consistieron en náusea y vómitos, alopecia, diarrea, fatiga, anorexia (disminución del apetito), inflamación de mucosa, cefalea, tos, erupción, dolor torácico, astenia, dolor laringofaríngeo (dolor orofaríngeo), y estreńimiento. En la experiencia postcomercialización, se han notificado casos de vasculitis cutánea en pacientes tratados con Neupogen. Se desconoce el mecanismo de la vasculitis en pacientes tratados con Neupogen. La frecuencia se estima como poco frecuente con base a los datos de los ensayos clínicos. Se han notificado casos de síndrome de Sweet (dermatosis febril aguda) en la experiencia postcomercialización. La frecuencia se estima como poco frecuente con base a los datos de los estudios clínicos. En ensayos clínicos y en la experiencia postcomercialización, se han notificado efectos adversos pulmonares incluyendo enfermedad pulmonar intersticial, edema pulmonar y casos de infiltración pulmonar resultando, en algunos casos, en insuficiencia respiratoria o síndrome de distrés respiratorio agudo (SDRA), que pueden llegar a ser fatales (ver Precauciones). Tras la administración de filgrastim se han notificado casos poco frecuentes de esplenomegalia y ruptura esplénica. Algunos casos de ruptura esplénica fueron fatales (ver Precauciones). En estudios clínicos y en la experiencia postcomercialización, se han notificado reacciones de hipersensibilidad incluyendo anafilaxia, erupción, urticaria, angioedema, disnea e hipotensión, que aparecieron al inicio o en la administración de dosis subsecuentes. En general, las notificaciones fueron más frecuentes tras la administración IV. En algunos casos, los síntomas han reaparecido tras la reexposición al fármaco, lo que sugiere la existencia de una relación causal. Debe suspenderse definitivamente el tratamiento con Neupogen en pacientes que desarrollen alguna reacción alérgica grave. En la experiencia postcomercialización, se han notificado casos aislados de crisis de células falciformes en pacientes con anemia de células falciformes o rasgos de anemia de células falciformes (ver Precauciones). La frecuencia se estima como poco frecuente con base a los datos de los ensayos clínicos. Se han notificado casos de pseudogota en pacientes con cáncer tratados con Neupogen. La frecuencia se estima como poco frecuente con base a los datos de los ensayos clínicos. Movilización de PBPC en donantes sanos: Se han notificado casos frecuentes pero generalmente asintomáticos de esplenomegalia y se han notificado casos poco frecuentes de ruptura esplénica en donantes sanos y en pacientes, tras la administración de filgrastim. Algunos casos de ruptura esplénica fueron fatales (ver Precauciones). Se han reportado reacciones adversas pulmonares (hemoptisis, hemorragia pulmonar, infiltración pulmonar, disnea e hipoxia) (ver Precauciones). La exacerbación de síntomas de artritis se ha observado con poca frecuencia. Se ha observado leucocitosis (leucocitos >50 x 109/l) en 41% de los donantes y trombocitopenia transitoria (plaquetas <100 x 109/l) en 35% de los donantes después de la administración de filgrastim y de los procesos de leucocitaféresis (ver Precauciones). En pacientes con NCG: Las reacciones adversas observadas incluyen esplenomegalia, que puede ser progresiva en una minoría de casos, ruptura esplénica y trombocitopenia (ver Precauciones). Las reacciones adversas, posiblemente relacionadas con el tratamiento con Neupogen y ocurriendo típicamente en menos del 2% de los pacientes con NCG, fueron reacción en el sitio de la inyección, cefalea, hepatomegalia, artralgia, alopecia, osteoporosis y erupción. Durante el uso a largo plazo se ha observado vasculitis cutánea en el 2% de los pacientes con NCG. En pacientes con VIH: La esplenomegalia se reportó relacionada al tratamiento con Neupogen en menos del 3% de los pacientes. En todos los casos, se consideró de leve a moderada durante la evaluación física y la evolución clínica fue benigna; a ninguno de los pacientes se le diagnosticó hiperesplenismo y ninguno debió someterse a una esplenectomía. Como la esplenomegalia es frecuente en los pacientes con infección por VIH y la mayoría de los pacientes con SIDA la presentan en una variedad de grados, no está clara su relación con el tratamiento con Neupogen (ver Precauciones). b.Población pediátrica: Datos de estudios clínicos en pacientes pediátricos indican que la seguridad y eficacia de Neupogen son similares en adultos y en nińos que reciben quimioterapia citotóxica, sugiriendo que la farmacocinética de filgrastim no cambia con la edad. La única reacción adversa consistentemente notificada fue el dolor músculo-esquelético, que no cambia respecto a la experiencia en la población adulta. No hay datos suficientes para evaluar el uso de Neupogen en pacientes pediátricos. b Otras poblaciones especiales: Uso geriátrico: No se han observado diferencias globales en la seguridad y eficacia en pacientes mayores de 65 ańos en comparación con pacientes adultos jóvenes (> 18 ańos de edad) que reciben quimioterapia citotóxica, y, en la experiencia clínica, no se han identificado diferencias entre las respuestas de los pacientes de edad avanzada y los adultos jóvenes. No hay datos suficientes para evaluar el uso de Neupogen en pacientes geriátricos en otras indicaciones aprobadas de Neupogen. Pacientes pediátricos con NCG: Se han notificado casos de densidad ósea disminuida y osteoporosis en pacientes pediátricos con neutropenia crónica grave que reciben tratamiento crónico con Neupogen. La frecuencia se estima como "frecuente" con base a los datos de los ensayos clínicos.
Contraindicaciones: Hipersensibilidad al principio activo o a alguno de los excipientes.
Advertencias: Neupogen no debe aplicarse para incrementar la dosis de la quimioterapia citotóxica más allá de los esquemas de dosificación establecidos. Neupogen no debe administrarse a pacientes con neutropenia congénita grave que desarrollen leucemia o tengan evidencia de evolución leucémica. Se ha reportado hipersensibilidad, incluyendo reacciones anafilácticas, en pacientes tratados con Neupogen, tanto con tratamientos iniciales o subsecuentes. Suspender permanentemente Neupogen en pacientes con hipersensibilidad clínica significativa. No administre Neupogen a pacientes con antecedentes de hipersensibilidad a filgrastim o pegfilgrastim. Como con todas las proteínas terapéuticas, existe potencial para inmunogenecidad. La frecuencia de generación de anticuerpos en contra de filgrastim es generalmente baja. Pueden producirse anticuerpos de unión como es esperado con todos los tratamientos biológicos, sin embargo, no se han asociado con actividad neutralizante.
Precauciones:Crecimiento de células malignas: El factor estimulante de colonias de granulocitos puede promover el crecimiento in vitro de las células mieloides y se han observado efectos similares en algunas células no mieloides in vitro. La seguridad y la eficacia de la administración de Neupogen en pacientes con síndromes mielodisplásicos o leucemia mieloide crónica no se han establecido. El uso de Neupogen no está indicado en estas enfermedades. Se debe poner atención especial para distinguir el diagnóstico de leucemia mieloide crónica en transformación blástica del de leucemia mieloide aguda. Debido a los pocos datos disponibles sobre la seguridad y eficacia en pacientes con leucemia mieloide aguda (LMA) secundaria, Neupogen debe administrarse con precaución. No se ha determinado la seguridad y eficacia de la administración de Neupogen en pacientes con LMA de novo menores de 55 ańos y con citogenética favorable (t(8;21), t(15;17) e inv(16)). Otras precauciones especiales: El monitoreo de la densidad ósea puede estar indicado en pacientes que presenten enfermedad osteoporótica de base y sean tratados con Neupogen durante más de 6 meses. Se han notificado efectos adversos pulmonares, en particular, enfermedad pulmonar intersticial, tras la administración de G-CSF. Pacientes con antecedente reciente de infiltrados pulmonares o neumonía pueden presentar mayor riesgo. La aparición de síntomas respiratorios tales como tos, fiebre y disnea, en asociación con signos radiológicos de infiltración y deterioro en la función pulmonar, pueden ser los síntomas preliminares del síndrome de distrés respiratorio agudo (SDRA). Se deberá suspender la administración de Neupogen y administrar el tratamiento apropiado. Se ha notificado síndrome de fuga capilar tras la administración de factores estimulantes de colonias de granulocitos, que se caracteriza por hipotensión, hipoalbuminemia, edema y hemoconcentración. Pacientes que desarrollen síntomas del síndrome de fuga capilar deben ser supervisados estrechamente y recibir tratamiento sintomático estándar, que puede incluir cuidados intensivos (ver Efectos colaterales). Ha habido reportes de glomerulonefritis en pacientes tratados con filgrastim y pegfilgrastim. En general, estos acontecimientos de glomerulonefritis se han resuelto posterior a la reducción de la dosis o suspensión del filgrastim y pegfilgrastim. Se recomienda la monitorización de análisis de orina. La cubierta de la aguja de la jeringa prellenada puede contener caucho natural (un derivado del látex) que puede provocar reacciones alérgicas. Precauciones especiales en pacientes con cáncer: Se han reportado casos poco frecuentes de esplenomegalia y ruptura esplénica tras la administración de filgrastim. Algunos casos de ruptura esplénica fueron fatales. Debe considerarse el diagnóstico de esplenomegalia o ruptura esplénica en pacientes tratados con filgrastim que reporten dolor en la parte superior izquierda del abdomen y/o dolor en el extremo del hombro. Leucocitosis: Cuentas leucocitarias de 100 x 109/l o superiores se han observado en menos del 5% de los pacientes recibiendo Neupogen en dosis superiores a 0.3 MU/kg/día (3 mcg/kg/día). No se ha observado ningún efecto adverso directamente atribuible a este grado de leucocitosis. Sin embargo, dada la posibilidad de que aparezcan reacciones asociadas con leucocitosis intensa, debe controlarse periódicamente la cuenta de leucocitos en intervalos regulares durante la terapia con Neupogen. Si la cuenta leucocitaria supera 50 x 109/l después del punto mínimo esperado, se debe suspender inmediatamente el tratamiento con Neupogen. Sin embargo, durante el período de administración de Neupogen para movilización de células progenitoras hematopoyéticas en sangre periférica, la administración de Neupogen debe suspenderse o reducir la dosis si la cuenta de leucocitos aumenta por arriba de 70 x 109/l. Riesgos asociados con el aumento de la dosis de quimioterapia: Se debe tener especial cuidado cuando se administran dosis altas de quimioterapia, ya que no se ha demostrado una mejoría en los resultados obtenidos sobre el tumor y la intensificación de la quimioterapia puede conducir a efectos de mayor toxicidad cardíaca, pulmonar, neurológica o dermatológica (por favor consulte la información para prescribir de los agentes quimioterapéuticos específicos utilizados). El tratamiento con Neupogen solo, no evita la trombocitopenia y anemia debidas a la quimioterapia mielosupresora. Pacientes tratados con quimioterapia en dosis altas (p.ej., dosis plenas del protocolo prescrito), pueden estar en mayor riesgo de trombocitopenia y anemia. Se recomienda vigilar periódicamente la cuenta plaquetaria y el valor hematocrito. Deben tomarse medidas de precaución especiales cuando se administren quimioterapéuticos como monoterapia o combinados que causan trombocitopenia grave. Se ha demostrado que el uso de células progenitoras hematopoyéticas en sangre periférica (PBPC's) movilizadas por Neupogen reduce la intensidad y duración de la trombocitopenia tras la quimioterapia mieloablativa o mielosupresora. Otras precauciones especiales: No se han estudiado los efectos de Neupogen en pacientes con disminución considerable de los progenitores mieloides. Neupogen actúa principalmente sobre los precursores de los neutrófilos, para ejercer sus efectos en elevar la cuenta de neutrófilos circulantes. Por eso, la respuesta al medicamento podría ser menor en los pacientes con disminución de las células precursoras (como aquellos sometidos a radioterapia o quimioterapia intensivas, o aquellos con infiltración neoplásica de la médula ósea). Se han reportado ocasionalmente trastornos vasculares, incluyendo enfermedad venoclusiva y alteraciones en el volumen de los fluidos, en pacientes sometidos a altas dosis de quimioterapia seguida de trasplante. Han sido reportados casos de enfermedad de injerto contra huésped (EICH) y muertes en pacientes que recibían G-CSF tras la realización de trasplante alogénico de médula ósea (ver Efectos colaterales y Farmacología). El aumento de la actividad hematopoyética de la médula ósea en respuesta a la terapia con factor de crecimiento, ha sido asociado con resultados anormales transitorios en escaneos óseos. Esto debe ser considerado cuando se interpreten resultados de imágenes óseas. Precauciones especiales en pacientes sometidos a movilización de PBPC: Movilización: No existen estudios comparativos, prospectivos, aleatorizados entre los 2 métodos de movilización recomendados (Neupogen solo, o en combinación con quimioterapia mielosupresora) dentro de la misma población de pacientes. Dada la variabilidad individual entre pacientes y la variabilidad interanalítica de las cuentas de células CD34+, resulta difícil establecer una comparación directa entre los diferentes estudios. Es difícil, por lo tanto, recomendar un método óptimo. La elección del método de movilización debe de realizarse de acuerdo con los objetivos del tratamiento para cada paciente en particular. Exposición previa a agentes citotóxicos: Los pacientes que han sido sometidos a una terapia mielosupresora previa intensiva, pueden no presentar una movilización suficiente de PBPC como para conseguir el rendimiento mínimo recomendado (≥2.0 x 106 células CD34+/ kg) o aceleración en la recuperación plaquetaria, en el mismo grado. Algunos agentes citotóxicos muestran toxicidad especial en el reservorio de células progenitoras hematopoyéticas y pueden afectar negativamente la movilización de las células progenitoras. Agentes tales como melfalán, carmustina (BCNU) y carboplatino cuando son administrados por períodos prolongados, antes de la movilización de las células progenitoras pueden reducir el rendimiento de este método. Sin embargo, si resulta eficaz para la movilización de células progenitoras hematopoyéticas la administración de melfalán, carboplatino o BCNU junto con Neupogen. Cuando se requiera efectuar trasplante de PBPC, se recomienda planificar el procedimiento de movilización de células madre al comienzo del período de tratamiento del paciente. En estos pacientes, antes de administrar altas dosis de quimioterapia, se prestará especial atención al número de células progenitoras movilizadas. Si los rendimientos no son adecuados, según el valor citado, se deben considerar otras formas alternativas de tratamiento que no requieran el soporte de células progenitoras. Valoración del rendimiento de células progenitoras: Se recomienda prestar especial atención al método de cuantificación para valorar el número de células progenitoras recolectadas en los pacientes tratados con Neupogen. Los resultados de los análisis de la citometría de flujo para determinar el número de células CD34+ varían en función de la precisión de la metodología usada, debiéndose interpretar con precaución las recomendaciones numéricas basadas en estudios realizados en otros laboratorios. Los análisis estadísticos de la relación entre el número de células CD34+ reinfundidas y la velocidad de recuperación plaquetaria tras altas dosis de quimioterapia indican que dicha relación es compleja pero continua. La recomendación de un rendimiento mínimo de ≥2.0 x 106 células CD34+/kg se basa en los datos publicados que consiguieron una recuperación hematológica suficiente. Los rendimientos superiores parecen estar en correlación con una recuperación más rápida, y los inferiores con una recuperación más lenta. Precauciones especiales en donantes sanos sometidos a movilización PBPC: La movilización de PBPC no ofrece ningún beneficio clínico directo para los donantes sanos y solamente debe considerarse en el marco de un trasplante alogénico de células madre. La movilización de PBPC solamente debe considerarse en donantes que cumplan los criterios de elegibilidad clínicos y de laboratorio para la donación de células madre prestando especial atención a los valores hematológicos y a las enfermedades infecciosas. La seguridad y eficacia de Neupogen en donantes sanos menores de 16 ańos o mayores de 60 ańos no se han evaluado. Se han reportado casos muy frecuentes de trombocitopenia en pacientes que reciben Neupogen. Por lo tanto, la cuenta de plaquetas debe monitorearse de manera cercana. Después de la administración de filgrastim y los procesos de leucocitaféresis se ha observado trombocitopenia transitoria (plaquetas < 100 x 109/l) en 35% de los pacientes estudiados. Entre ellos, se reportaron dos casos con plaquetas < 50 x 109/l que se atribuyeron al procedimiento de leucocitaféresis. En caso de ser necesaria más de una leucocitaféresis, se debe prestar especial atención a los donantes que antes de la leucocitaféresis tengan plaquetas < 100 x 109/l; en general no se recomienda hacer aféresis si las plaquetas están por debajo de 75 x 109/l. No deben realizarse leucocitaféresis a donantes tratados con anticoagulantes o con trastornos hemostáticos. Debe suspenderse la administración de Neupogen o reducirse la dosis si la cuenta de leucocitos supera los 70 x 109/l. Los donantes que reciben factores estimulantes de las colonias de granulocitos (G CSF) para la movilización de PBPC deben ser vigilados estrechamente hasta que sus valores hematológicos regresen a la normalidad. En donantes sanos se han observado alteraciones citogenéticas transitorias después de recibir tratamiento con G-CSF. Se desconoce la trascendencia de estos cambios. Aun así, no se puede descartar el riesgo de estimulación de alguna clona mieloide maligna. Se recomienda que el centro de aféresis lleve un control y seguimiento sistemático de los donantes de células madre durante al menos 10 ańos para asegurar el monitoreo de la seguridad a largo plazo. Se han reportado casos frecuentes pero generalmente asintomáticos de esplenomegalia y casos poco frecuentes de ruptura esplénica en donadores sanos (y pacientes), tras la administración de G-CSF's. Algunos casos de ruptura esplénica fueron mortales. Por lo tanto, debe realizarse una cuidadosa monitorización clínica del tamańo del bazo (p. ej. examen clínico, ultrasonido). Debe considerarse el diagnóstico de ruptura esplénica en los donadores y/o pacientes que refieran dolor abdominal superior izquierdo o en el extremo del hombro. En donadores sanos, se han reportado casos frecuentes de disnea y poco frecuentes de otras reacciones adversas pulmonares (hemoptisis, hemorragia pulmonar, infiltración pulmonar e hipoxia). En caso de sospecha o confirmación de reacciones adversas pulmonares, debería considerarse suspender el tratamiento con Neupogen y administrar el cuidado médico apropiado. Precauciones especiales en los receptores de células progenitoras hematopoyéticas periféricas alogénicas (PBPC's) movilizadas con Neupogen: Los datos disponibles indican que las interacciones inmunitarias entre células progenitoras hematopoyéticas periféricas alógenas (PBPC) trasplantadas y el receptor pueden asociarse a un aumento del riesgo de enfermedad aguda o crónica del injerto contra huésped (EICH) en comparación con el trasplante de médula ósea. Precauciones especiales en pacientes con NCG: Biometría hemática: Se han notificado casos frecuentes de trombocitopenia en pacientes tratados con Neupogen. La cuenta plaquetaria debe controlarse cuidadosamente, sobre todo durante las primeras semanas de tratamiento con Neupogen. En pacientes que desarrollen trombocitopenia, es decir, en aquellos con una cuenta de plaquetas persistentemente < 100.000/mm3 debe valorarse la posibilidad de suspender el tratamiento con Neupogen de forma intermitente o, al menos, reducir la dosis. Existen también otros cambios en la biometría hemática como la anemia y el aumento transitorio de los progenitores mieloides que obligan a vigilar cuidadosamente la cuenta celular. Transformación hacia leucemia o síndrome mielodisplásico: Se debe establecer cuidadosamente el diagnóstico de NCG y diferenciarlo de otros trastornos hematopoyéticos como la anemia aplásica, mielodisplasia y leucemia mieloide. Antes del tratamiento debe realizarse una biometría hemática completa con fórmula leucocitaria y cuenta de plaquetas, así como un estudio morfológico de la médula ósea y cariotipo. Hubo una frecuencia baja (aproximadamente 3%) de síndrome mielodisplásico (SMD) o leucemia en pacientes con NCG incluidos en ensayos clínicos y tratados con Neupogen. Esta observación sólo se ha hecho en pacientes con neutropenia congénita. El SMD y las leucemias son complicaciones naturales de la enfermedad y su relación con el tratamiento de Neupogen es incierta. Un subgrupo de aproximadamente 12% de pacientes cuyas evaluaciones citogenéticas fueron normales a nivel basal, presentaron posteriormente anormalidades, incluyendo monosomía 7, en la evaluación repetida de rutina. No está claro en la actualidad si el tratamiento mantenido de los pacientes con NCG predispone a éstos hacia anormalidades citogenéticas, SMD o transformación leucémica. Se recomienda efectuar a los pacientes exámenes morfológicos y citogénicos de la médula ósea a intervalos regulares (aproximadamente cada 12 meses). Otras precauciones especiales: Se deben excluir las causas que provoquen neutropenia transitoria, como es el caso de las infecciones virales. Tras la administración de filgrastim se han reportado casos muy frecuentes de esplenomegalia, y casos frecuentes de ruptura esplénica. Debe considerarse el diagnóstico de esplenomegalia o ruptura esplénica en pacientes tratados con filgrastim que refieran dolor en la parte superior izquierda del abdomen o dolor en el extremo del hombro. La esplenomegalia es una consecuencia directa del tratamiento con Neupogen. Treinta y uno por ciento (31%) de los pacientes incluidos en estudios clínicos presentaron esplenomegalia detectable por palpación. El aumento en el volumen del bazo, medido radiográficamente, se presentó al comienzo del tratamiento con Neupogen y tendió a estabilizarse. La progresión de la esplenomegalia disminuyó o se detuvo al reducir la dosis, y sólo en 3% de los pacientes se requirió esplenectomía. Se debe evaluar de forma regular el tamańo del bazo. Para detectar un aumento anómalo del volumen esplénico basta con realizar palpación abdominal. Hematuria fue reportada de manera frecuente y proteinuria ocurrió en un pequeńo número de casos. Es necesario efectuar un análisis regular de orina para controlar estos acontecimientos. La seguridad y la eficacia no están establecidas en recién nacidos y en pacientes con neutropenia autoinmune. Precauciones especiales en pacientes con infección por VIH: Se han notificado de manera frecuente casos de esplenomegalia tras la administración de Neupogen. Debe considerarse un diagnóstico de esplenomegalia o ruptura esplénica en individuos tratados con filgrastim que refieran dolor en la parte superior izquierda del abdomen o en el extremo del hombro. Biometría hemática: La CAN debe monitorizarse cuidadosamente, especialmente durante las primeras semanas de tratamiento con Neupogen. Algunos pacientes responden rápidamente a la dosis inicial de Neupogen con un aumento considerable en la cuenta de neutrófilos. Se recomienda la evaluación diaria de la CAN durante los 2-3 primeros días de la administración de Neupogen. Después, se recomienda que la CAN se evalúe al menos 2 veces por semana durante las 2 primeras semanas y posteriormente, 1 vez a la semana o 1 vez cada 2 semanas durante la terapia de mantenimiento. Durante la administración intermitente de 30 MU (300 mcg)/día de Neupogen pueden producirse amplias fluctuaciones en la CAN a lo largo del tiempo. Para determinar la cifra mínima de neutrófilos, se recomienda que se tomen muestras sanguíneas para medir la CAN inmediatamente antes de cualquiera de las dosis pautadas de Neupogen. Riesgos asociados con dosis más altas de medicamentos mielosupresores: El tratamiento con Neupogen solo, no descarta la trombocitopenia ni la anemia causada por los medicamentos mielosupresores. Como resultado de la posibilidad de recibir dosis mayores o un número mayor de estos medicamentos con el tratamiento con Neupogen, el riesgo de trombocitopenia y anemia puede ser mayor. Se recomienda monitorear la biometría hemática periódicamente (ver arriba). Mielodepresión de causa infecciosa o neoplásica: La neutropenia puede deberse a infecciones oportunistas tales como el complejo Mycobacterium avium o a neoplasias tales como linfomas malignos que infiltran la médula ósea. En los pacientes que se conoce tienen infiltraciones infecciosas en la médula ósea o malignidad, se debe considerar la administración de un tratamiento adecuado para dichas condiciones, además de la administración de Neupogen para el tratamiento de la neutropenia. El efecto de Neupogen sobre la neutropenia causada por tumores o por infecciones con infiltración de la médula ósea, no está bien establecido. Precauciones especiales en pacientes con rasgo de células falciformes o anemia de células falciformes: Se han notificado crisis de anemia de células falciformes, en algunos casos fatales, en pacientes con rasgo de células falciformes o anemia de células falciformes a los que se les ha administrado Neupogen. El médico deberá actuar con precaución al considerar la administración de Neupogen en pacientes con rasgo de células falciformes o anemia de células falciformes. En todos los pacientes: Neupogen contiene sorbitol (E420). Los pacientes con problemas hereditarios raros de intolerancia a la fructosa no deben utilizar este medicamento. Neupogen contiene menos de 1 mmol (23 mg) de sodio por 0.6 mg/ml, por lo que se considera esencialmente libre de sodio. Para mejorar la trazabilidad de los G-CSF's, debería registrarse claramente en el expediente del paciente la marca comercial del producto administrado.
Interacciones Medicamentosas: No se han establecido definitivamente la seguridad y eficacia de Neupogen, administrado el mismo día que la quimioterapia citotóxica mielosupresora. No se recomienda el uso de Neupogen entre las 24 horas previas y las 24 horas posteriores a la quimioterapia, debido a que las células mieloides en fase de replicación rápida son muy sensibles a la quimioterapia citotóxica mielosupresora. Los datos preliminares provenientes de un pequeńo número de pacientes tratados simultáneamente con Neupogen y 5-fluorouracilo indican que se puede exacerbar la gravedad de la neutropenia. Todavía no se ha investigado en ensayos clínicos la posible interacción con otros factores de crecimiento hematopoyético y citocinas. Debido a que el litio estimula la liberación de neutrófilos, es probable que potencie el efecto de Neupogen. Aunque esta interacción no se ha investigado formalmente, no hay evidencia de que pueda ser nociva.
Sobredosificación:Sobredosis y tratamiento: El efecto de la sobredosis de Neupogen no se ha establecido. La interrupción del tratamiento de Neupogen se acompańa, habitualmente, de una disminución del 50% de los neutrófilos circulantes al cabo de 1 a 2 días y de una normalización al cabo de 1 a 7 días.
Incompatibilidades: Neupogen no debe diluirse con soluciones salinas. El filgrastim diluido, puede adsorberse al vidrio y materiales plásticos. Este medicamento no debe mezclarse con otros productos excepto con los mencionados en Posología.
Conservación: Almacenar a una temperatura entre 2şC y 8ş C. Sin congelar. Para el producto diluido en solución de glucosa 5%, almacenar entre 2ş C y 8ş C por 24 horas, bajo condiciones asépticas validadas y controladas.
Presentaciones: Caja con 1 jeringa prellenada con 30 MU/0.5 ml de Neupogen solución inyectable. Las jeringas prellenadas están hechas de vidrio tipo I y tienen una aguja de acero inoxidable permanentemente incorporada en la punta. La cubierta de la aguja de la jeringa prellenada contiene caucho natural (un derivado del látex) o gaucho sintético, (ver Precauciones). Caja con 1 frasco-ampolla con 30 MU/1 ml de Neupogen solución inyectable. Los frascos-ampolla son hechos de vidrio tipo I con tapón de goma.
 Laboratorio
Laboratorio