Composición: Ingrediente activo: Temsirolimus. Excipientes: Concentrado de Temsirolimus para Inyección, 25 mg/ml. Ingredientes inactivos: Alcohol Deshidratado [Etanol Anhidro], Dl-alfa-tocoferol, Propilenglicol y Ácido Cítrico Anhidro, Nitrógeno. Diluyente para concentrado de Temsirolimus para Inyección. Ingredientes inactivos: Polisorbato 80, Nitrógeno, 400 (Macrogol 400), Alcohol Deshidratado [Etanol Anhidro].
Indicaciones: Temsirolimus concentrado inyectable está indicado para el tratamiento de primera línea de pacientes con carcinoma avanzado de células renales que presenten como mínimo 3 a 6 factores de riesgo pronósticos.
Posología:Modo de administración: Intravenosa: Preparación y precauciones para la administración: La dosis recomendada de temsirolimus concentrado Inyectable para el carcinoma avanzado de células renales es de 25 mg, perfundida durante un período de 30 a 60 minutos 1 vez por semana. El tratamiento deberá continuar hasta que el paciente ya no demuestre beneficios clínicos con el tratamiento o hasta que se produzca toxicidad inaceptable. No es necesario realizar modificaciones posológicas especiales en ninguna de las poblaciones estudiadas (por ejemplo, por sexo, ancianos). El tratamiento de las reacciones sospechadas al medicamento podrá requerir la interrupción transitoria del tratamiento y/o una reducción de la dosis de temsirolimus. En caso de que una reacción sospechada no pudiera ser tratada con la postergación de la dosis, se podrá reducir la dosis de temsirolimus concentrado Inyectable en disminuciones de 5 mg/semana. Para carcinoma de células renales: Instrucciones para la administración endovenosa: Durante el manejo y la preparación de las mezclas, temsirolimus concentrado Inyectable deberá protegerse de las luces solares y artificiales excesivas. Temsirolimus concentrado Inyectable debe inspeccionarse visualmente para detectar partículas y decoloración antes de la administración, cuando la solución y el envase lo permitan. Las bolsas/envases que entren en contacto con temsirolimus concentrado inyectable deben ser de vidrio, poliolefina o polietileno. Si se encuentra material particulado en el vial no utilizarlo. Usar un nuevo vial. Premedicación: Los pacientes deberán recibir medicación profiláctica de 25 a 50 mg de difenhidramina endovenosa (o equivalente) aproximadamente 30 minutos antes de comenzar la infusión de cada dosis de temsirolimus. Deberá suspenderse la infusión en caso de manifestarse una reacción de hipersensibilidad durante la infusión de temsirolimus. Una vez resuelta y después de observar al paciente por al menos 30 a 60 minutos y a criterio del médico, se podrá reanudar el tratamiento con la administración de un antagonista del receptor H1 (o equivalente), si no se hubiera administrado anteriormente, y/o un antagonista del receptor H2 (tales como famotidina intravenosa 20 mg o ranitidina intravenosa 50 mg) aproximadamente 30 minutos antes de reiniciar la infusión de temsirolimus. La infusión podrá entonces reanudarse a menor velocidad (hasta 60 minutos). Dilución:Cada vial de temsirolimus se debe diluir con diluyente de acuerdo con las siguientes instrucciones: Los contenidos diluidos requeridos de cada vial se deben combinar en una jeringa de inyección con 250 ml, de 0.9% de cloruro de sodio. La solución diluida (concentrado y diluyente) se debe inspeccionar visualmente para detectar material particulado y decoloración. Al preparar la solución de administración de temsirolimus, seguir el siguiente proceso de dilución de 2 pasos en forma aséptica:Paso 1: Inyectar 1.8 ml del diluyente para temsirolimus concentrado inyectable en el frasco-ampolla de temsirolimus concentrado inyectable. Mezclar bien invirtiendo el frasco-ampolla. La concentración de la droga será de 10 mg/ml. Asegurarse de dejar pasar tiempo suficiente para que las burbujas de aire desaparezcan. La solución es clara a ligeramente turbia, incolora a amarillo claro o amarillo y esencialmente libre de partículas visuales. Un volumen de 1.2 ml del concentrado contiene un total de 30 mg de producto farmacéutico. Cuando se combinan 1.2 ml del concentrado con 1.8 ml del diluyente, se obtiene un volumen total de 3.0 ml. Treinta miligramos (30 mg) del medicamento por 3.0 ml = 10 mg/ml de producto medicinal. La mezcla de concentrado farmacéutico-diluyente es estable durante 24 horas a temperatura ambiente controlada de 20ş a 25şC. Paso 2: Retirar la cantidad necesaria de la mezcla de temsirolimus concentrado inyectable/diluyente preparada en el paso 1 (10 mg/ml) e inyectarla rápidamente en 250 ml de solución parenteral de cloruro de sodio al 0.9% para asegurar un mezclado adecuado. Mezclar la mezcla invirtiendo la bolsa o frasco. No agitar demasiado para evitar la formación de espuma. Se debe inspeccionar visualmente la solución diluida final en la bolsa o matraz para detectar material particulado. Administración: La administración de la solución final diluida para infusión debe completarse dentro de las 6 horas desde el momento en que la mezcla del concentrado/diluyente se agrega a la solución parenteral de cloruro de sodio (0.9%). Los materiales adecuados de administración deben ser de vidrio, poliolefina o polietileno para evitar la pérdida excesiva del medicamento y para reducir la velocidad de extracción de di-(2-etilhexil)ftalato (DEHP). Los materiales de administración deben componerse de una tubuladura libre de DEHP y de cloruro de polivinilo (PVC) con el filtro adecuado. Para la administración se recomienda un filtro de polietersulfona en línea con un tamańo de poro que no supere los 5 micrones, para evitar la posibilidad de que se infundan partículas de más de 5 micrones. Si el sistema de administración disponible no tiene un filtro en línea incorporado, se debe ańadir un filtro al final de éste (es decir, un filtro final) antes de que el preparado llegue a la vena del paciente. Se pueden usar diferentes filtros finales, con tamańos de poros de entre 0.2 micrones hasta 5 micrones. No se recomienda el uso de un filtro en línea y un filtro final juntos. Es importante seguir estrictamente las recomendaciones indicadas en dosis y vía de administración. Temsirolimus concentrado inyectable, una vez reconstituido, contiene polisorbato 80 que aumenta la velocidad de extracción de di-(2-etilhexil)ftalato (DEHP) del cloruro de polivinilo (PVC). Esto debe tenerse en cuenta durante la preparación y administración de temsirolimus concentrado inyectable, así como también el tiempo de conservación transcurrido en un envase de PVC después de la reconstitución. Es importante seguir estrictamente las recomendaciones indicadas en dosis y vía de administración. Carcinoma de células renales: La dosis recomendada de temsirolimus concentrado inyectable en el carcinoma de células renales avanzado es de 25 mg en infusión durante un periodo de 30 a 60 minutos 1 vez por semana. El manejo de la sospecha de reacciones a la droga podría requerir la interrupción temporal y/o la reducción temporal de la dosis de temsirolimus. Si sospecha de una reacción no es aconsejable el retraso de la dosis, luego temsirolimus concentrado inyectable se puede reducir en decrementos de 5 mg por semana. Uso en pacientes con disfunción renal: Después de una dosis endovenosa de 25 mg de temsirolimus marcado con [14C] en sujetos sanos, la eliminación renal de la radioactividad total fue del 4.6% de la dosis administrada. La eliminación renal es una vía secundaria, por lo tanto, no es de esperar que el deterioro renal influya marcadamente en la exposición a la droga, no siendo necesario ajustar la dosis de temsirolimus concentrado inyectable en pacientes con insuficiencia renal. No se han llevado a cabo estudios en pacientes con diversos grados de disfunción renal. No se ha evaluado el tratamiento con temsirolimus concentrado inyectable en pacientes sometidos a hemodiálisis. Uso en pacientes con dańo hepático: La seguridad y la farmacocinética de temsirolimus se evaluaron en un estudio de Fase 1 con escalamiento de la dosis en 110 pacientes con cáncer con función hepática normal o con grados variables de insuficiencia hepática. Los pacientes con bilirrubina > 1.5 x ULN al basal experimentaron una toxicidad mayor que los pacientes con bilirrubina ≤1.5 x ULN al basal durante el tratamiento con temsirolimus. La frecuencia global de reacciones adversas de grado ≥3 y muertes, incluyendo las muertes por enfermedad progresiva, fueron mayores en pacientes con bilirrubina > 1.5 x ULN al basal. El temsirolimus está contraindicado en pacientes con bilirrubina > 1.5 x ULN, debido al riesgo aumentado de muerte, incluidas las muertes debido a la progresión del cáncer subyacente. Tenga precaución al tratar a pacientes con insuficiencia hepática leve. Las concentraciones de temsirolimus y su metabolito sirolimus se incrementaron en los pacientes con elevación de los niveles de AST o de bilirrubina. Se recomienda evaluar los niveles de AST y de bilirrubina antes de la iniciación de temsirolimus y posteriormente de forma periódica. Uso en nińos: La información disponible sobre el uso de temsirolimus en pacientes pediátricos es limitada. No se ha establecido la seguridad y eficacia de temsirolimus en pacientes pediátricos con tumores sólidos recurrentes/refractarios. Uso en pacientes ancianos: No se recomiendan ajustes específicos de la dosis en los ancianos.
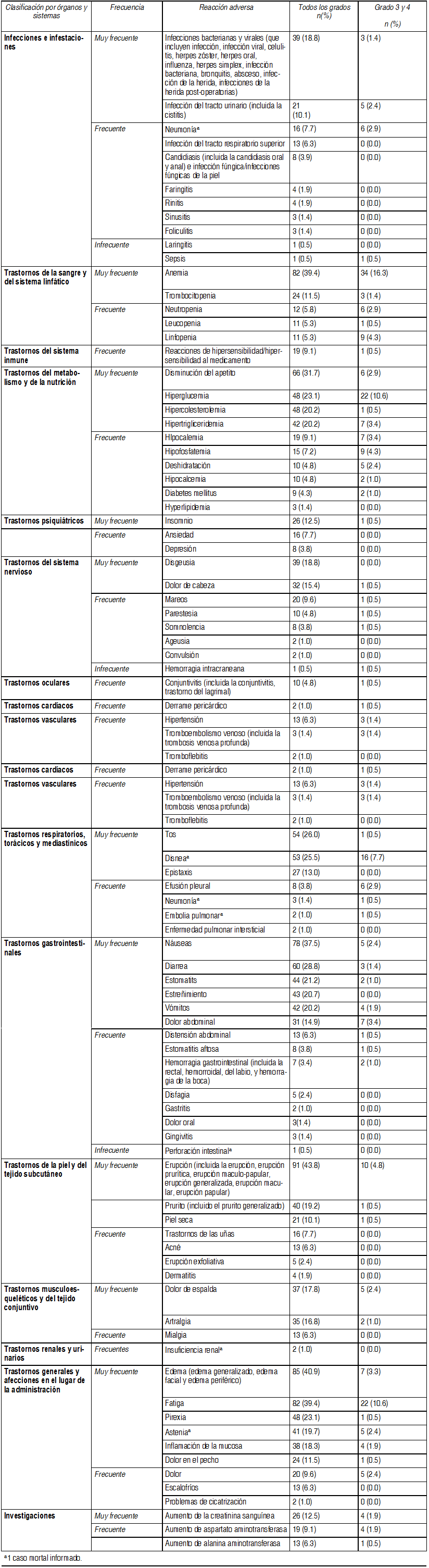
Efectos Colaterales:La frecuencia esperada de reacciones adversas se presenta en las categorías de frecuencia de CIOMS:Muy frecuente: ≥10%. Frecuente: ≥1% y <10% Infrecuentes: ≥0.1% y <1%. Raras: ≥0.01% y <0.1%. Muy raras: <0.01%.
Las reacciones adversas serias observadas en los ensayos clínicos de temsirolimus para el carcinoma de células renales, incluyen: anafilaxis, alteración de la cicatrización de heridas, insuficiencia renal con desenlace fatal, derrame pericárdico (incluyendo derrames pericárdicos hemodinámicamente significativos que requieren una intervención) convulsiones, y embolia pulmonar, y embolia pulmonar. Post-comercialización y otras experiencias clínicas:Clasificación por órganos y sistemas/Frecuencia/Reacciones adversas: Trastornos del sistema inmune: No conocida*: Reacciones de tipo edema angioneurótico. Trastornos de la piel y del tejido subcutáneo: No conocida: Síndrome de Stevens-Johnson. Trastornos musculoesqueléticos y del tejido conjuntivo: No conocida: Rabdomiólisis. Infecciones e infestaciones: Rara: Neumonía por Pneumocystis jiroveci. *no se puede calcular a partir de los datos disponibles. Reacciones de tipo edema angioneurótico en algunos pacientes que recibieron temsirolimus e inhibidores de ACE simultáneamente. Se han informado casos de neumonía por Pneumocystis jiroveci, algunos con resultados mortales.
Contraindicaciones: Temsirolimus endovenoso está contraindicado en pacientes con hipersensibilidad conocida al temsirolimus o a alguno de los componentes de esta formulación. Temsirolimus está contraindicado en pacientes con bilirrubina >1.5 x ULN.
Advertencias:Reacciones de hipersensibilidad a la infusión: Se han observado reacciones de hipersensibilidad/a la infusión, (que incluyeron algunas reacciones con riesgo de vida y raros casos mortales) , incluye pero no se limita a, rubefacción, dolor toráxico, disnea, hipotensión, apnea, pérdida del conocimiento, hipersensibilidad y anafilaxia asociadas con la administración de temsirolimus. Estas reacciones pueden aparecer al comienzo de la primera infusión, pero también con las infusiones subsiguientes. Se recomienda monitoreo del paciente al iniciarse y durante la infusión, junto con tratamiento de apoyo adecuado disponible. La infusión de temsirolimus deberá interrumpirse en todo paciente que presente una reacción grave a la infusión y administrarse el tratamiento médico adecuado. Deberá evaluarse la relación beneficio/riesgo antes de continuar el tratamiento con temsirolimus en los pacientes con reacciones graves o riesgosas para la vida. Sirolimus es el metabolito principal de temsirolimus; por lo tanto, temsirolimus deberá administrarse con precaución en pacientes con conocida hipersensibilidad al sirolimus. Debido a que se recomienda administrar un antihistamínico H1 a los pacientes antes de comenzar la infusión endovenosa de temsirolimus, deberá emplearse con precaución en pacientes con hipersensibilidad conocida a un antihistamínico o en pacientes que no pueden recibir antihistamínicos por otros motivos médicos. Si un paciente presentara una reacción de hipersensibilidad durante la infusión de temsirolimus a pesar de la premedicación, deberá suspenderse la infusión y observarse al paciente durante por lo menos 30 a 60 minutos (según la severidad de la reacción). A criterio del médico, se podrá reanudar el tratamiento con la administración de un antagonista del receptor H1 (por ejemplo, difenhidramina), si no se hubiera administrado anteriormente, y/o un antagonista del receptor H2 (tales como famotidina intravenosa 20 mg o ranitidina intravenosa 50 mg) aproximadamente 30 minutos antes de reiniciar la infusión de temsirolimus. La infusión podrá entonces reanudarse a menor velocidad (hasta 60 minutos). Dańo hepático: El temsirolimus fue evaluado en un estudio de Fase 1 con escalamiento de la dosis estudio en 110 pacientes con función hepática normal o grados variables de insuficiencia hepática según lo definido por los niveles de AST y bilirrubina y en pacientes con trasplante hepático (Tabla 1). Durante el estudio, los pacientes con insuficiencia hepática grave y moderada presentaron una tasa aumentada de eventos adversos y muertes, incluidas las muertes debido a la progresión del cáncer subyacente (Tabla 1). Función hepática* /Rango de dosificación de temsirolimus (mg)/Eventos adversos grado ≥3** n (%)/Muertes totales*** n (%): Normal (n=25): 25 - 175: 20 (80,0): 2 (8.0). Leve (n=39):10 - 25: 32 (82.1): 5 (12.8). Moderada (n=20): 10 - 25: 19 (95.0): 8 (40.0). Grave (n=24): 7.5 - 15: 23 (95.8): 13 (54.2). Trasplante hepático (n=2): 10: 1 (50.0): 0 (0). *Grupos por función hepática: normal = bilirrubina y AST ≤ LMN; leve = bilirrubina > 1 - 1.5 × LMN o AST > LMN pero bilirrubina ≤ LMN; moderada = bilirrubina > 1.5 - 3 × LMN; grave = bilirrubina > 3 × LMN; trasplante hepático = cualquier bilirrubina y AST. ** Criterio de terminología común para eventos adversos, versión 3.0, lo que incluye todas las causas.*** Incluye las muertes debido al avance del cáncer subyacente y las reacciones adversas. La seguridad y la farmacocinética de temsirolimus se evaluaron en un estudio de Fase 1 con escalamiento de la dosis en 110 pacientes con cáncer con función hepática normal o con grados variables de insuficiencia hepática. Los pacientes con bilirrubina > 1.5 x ULN al basal experimentaron una toxicidad mayor que los pacientes con bilirrubina ≤ 1.5 x ULN al basal durante el tratamiento con temsirolimus. La frecuencia global de reacciones adversas de grado ≥ 3 y muertes, incluyendo las muertes por enfermedad progresiva, fueron mayores en los pacientes con bilirrubina > 1.5 x ULN al basal. El temsirolimus está contraindicado en pacientes con bilirrubina > 1.5 x ULN, debido al riesgo aumentado de muerte, incluidas las muertes debido a la progresión del cáncer subyacente. Tenga cuidado al tratar a pacientes con insuficiencia hepática leve. Las concentraciones de temsirolimus y su metabolito sirolimus se incrementaron en los pacientes con elevación de los niveles de AST o de bilirrubina. Se recomienda evaluar los niveles de AST y de bilirrubina antes de la iniciación de temsirolimus y posteriormente de forma periódica. Hiperglucemia/Intolerancia a la glucosa: El empleo de temsirolimus concentrado inyectable en pacientes con carcinoma renal estuvo asociado con elevaciones de la glucosa sérica. En el Estudio 1, un ensayo clínico de Fase 3 para el carcinoma de células renales (Estudio 3066K1-304), el 26% de los pacientes informó la hiperglucemia como un evento adverso. En el estudio 2, un ensayo clínico de fase 3 para el linfoma de células del manto (Estudio 3066K1-305), el 11% de los pacientes informó la hiperglucemia como un evento adverso. Esto puede originar la necesidad de un aumento en la dosis de insulina o el inicio del tratamiento con insulina y/o un hipoglucemiante oral. Se deberá indicar a los pacientes que informen si tienen mucha sed o mayor volumen o frecuencia de micción. Infecciones: Deberá observarse cuidadosamente a los pacientes con inmunosupresión por la aparición de infecciones, incluidas infecciones oportunistas. Se han informado casos de neumonía por Pneumocystis jiroveci (PJP), algunos con resultados mortales, en pacientes que recibieron temsirolimus, muchos de los cuales también recibieron corticosteroides u otros agentes inmunodepresivos. Para los pacientes que requieren el uso simultáneo de corticosteroides u otros agentes inmunodepresivos, se puede considerar la profilaxis de PJP. Enfermedad pulmonar intersticial: Se han comunicado casos de neumonitis intersticial inespecífica, que incluyeron raros casos fatales, en pacientes que recibieron temsirolimus endovenoso en forma semanal. Algunos pacientes no presentaban síntomas con neumonitis detectada por tomografía computada o radiografía de tórax. Otros presentaron síntomas tales como disnea, tos y fiebre. Algunos pacientes requirieron la suspensión de temsirolimus o tratamiento con corticoides y/o antibióticos mientras que otros continuaron con el tratamiento sin necesidad de intervención alguna. Se recomienda que los pacientes se sometan a una evaluación radiográfica basal mediante tomografía computarizada de pulmón o radiografía de tórax antes de la iniciación de la terapia con temsirolimus. Continúe con estas evaluaciones periódicamente, incluso en ausencia de síntomas clínicos respiratorios. Se recomienda seguimiento de los pacientes para detectar síntomas respiratorios clínicos. Si se desarrollan síntomas respiratorios clínicos significativos, tenga en cuenta la suspensión de la administración de temsirolimus hasta después de la recuperación de los síntomas y la mejora de los hallazgos radiográficos relacionados con neumonitis. Las infecciones oportunistas, tales como la neumonía por Pneumocystis jiroveci (PJP), se deberían considerar en los diagnósticos diferenciales. Se puede considerar el tratamiento empírico con corticoides y / o antibióticos. Para los pacientes que requieren el uso de corticosteroides, se puede considerar la profilaxis de PJP. Hiperlipidemia: El empleo de temsirolimus en pacientes con carcinoma de células renales estuvo asociado con elevación de los triglicéridos y colesterol séricos. En el Estudio 1, se informó hiperlipemia como un evento adverso en el 27% de los pacientes. En el Estudio 2, se informó hiperlipemia como un evento adverso en el 9.3% de los pacientes. Esto podría necesitar inicio o aumento de la dosis de hipolipidemiantes. Se recomienda controlar los triglicéridos y el colesterol en suero antes y durante el tratamiento con temsirolimus. Perforación intestinal: Se han registrado casos de perforación intestinal (incluidos casos fatales) en pacientes que recibieron temsirolimus. Complicaciones en cicatrización de heridas: El empleo de temsirolimus ha sido asociado con cicatrización anormal de heridas. Por lo tanto, se recomienda precaución al administrar temsirolimus en el período perioperatorio. Hemorragia intracerebral: Los pacientes con tumores del sistema nervioso central (tumores primarios o metástasis del SNC) y/o que reciban tratamiento anticoagulante pueden estar expuestos a un mayor riesgo de desarrollar hemorragia intracerebral (y hasta desenlace fatal) mientras reciben tratamiento con temsirolimus. Insuficiencia renal: Se ha observado insuficiencia renal (incluso casos fatales) en pacientes tratados con temsirolimus para el cáncer avanzado de células renales y/o con insuficiencia renal preexistente. Empleo concomitante de temsirolimus con sunitinib: La combinación de temsirolimus con sunitinib produjo toxicidad dosis-dependiente. Se observaron toxicidades dosis dependientes (rash maculopapuloso y eritematoso grado 3, gota/celulitis que requirieron hospitalización) en 2 de 3 pacientes tratados en el primer cohorte de un estudio de fase I con dosis de 15 mg intravenosos de temsirolimus por semana y 25 mg orales de sunitinib por día (1-28 días seguidos de un descanso de 2 semanas). Empleo concomitante de inhibidores de la enzima convertidora de angiotensina (ECA): Se han observado reacciones de tipo edema angioneurótico (incluyendo reacciones tardías que ocurren dos meses después de iniciada la terapia) en algunos pacientes tratados con temsirolimus e inhibidores ECA en forma concomitante. Ancianos: No se observaron diferencias generales específicas en cuanto a la seguridad entre los pacientes menores de 65 ańos y aquellos mayores de 65. De acuerdo con los resultados de un estudio de fase 3 en carcinoma de células renales, los pacientes ancianos pueden presentar mayor probabilidad de manifestar determinadas reacciones adversas, tales como edema, diarrea y neumonía. La sobrevida general en un grupo de pacientes de 65 ańos o más (n=64) tratados con temsirolimus en el estudio 1 fue más corta que aquella observada en los pacientes menores de 65 ańos. La importancia clínica del análisis de este subgrupo no es clara. No se recomienda ningún ajuste específico de la dosis para los pacientes mayores. Población pediátrica: No se recomienda el uso de temsirolimus en los pacientes pediátricos debido a la insuficiencia de datos respecto a su eficacia. Cataratas: Se ha observado cataratas en algunos pacientes que recibieron la combinación de temsirolimus e interferón alfa. Agentes inductores del metabolismo de CYP3A: Los agentes tales como carbamazepina, fenitoína, barbitúricos, rifabutina, rifampicina y la hierba de San Juan son fuertes inhibidores de CYP3A4/5 y pueden reducir la exposición al compuesto de los grupos activos, temsirolimus y su metabolito sirolimus. Por lo tanto, para pacientes con carcinoma de células renales, no deberá administrarse tratamiento concomitante con agentes que tengan potencial de inducción de CYP3A4/5. Si no se pudiera administrar un tratamiento alternativo, 1 dosis intravenosa semanal de 50 mg debe ser considerada. Agentes inhibidores del metabolismo de CYP3A: Los agentes tales como los inhibidores de las proteasas, antimicóticos, antibióticos macrólidos, nefazodona e inhibidores selectivos de la serotonina son fuertes inhibidores de CYP3A4 y pueden aumentar las concentraciones sanguíneas de los grupos activos, temsirolimus y su metabolito sirolimus. Por lo tanto, no deberá administrarse tratamiento concomitante con agentes que tengan potencial de inhibición de CYP3A4. El tratamiento concomitante con inhibidores moderados de CYP3A4 debe ser únicamente administrado con precaución en pacientes tratados con 25 mg y no debe administrarse a pacientes que reciban dosis superiores a 25 mg de temsirolimus. Deberán considerarse tratamientos alternativos que no tengan potencial de inhibición de CYP3A4. Vacunas: Deberá evitarse el empleo de vacunas atenuadas, tales como la triple viral (MMR), antipoliomielítica oral, BCG, vacuna contra la fiebre amarilla, contra la varicela y antitifoidea TY21a durante el tratamiento con temsirolimus.
Precauciones:Embarazo: No se han llevado a cabo estudios adecuados y bien controlados en mujeres embarazadas con temsirolimus. En estudios de toxicidad en animales con ratas y conejos, se observó aumento de la mortalidad embrio/fetal y disminución del desarrollo fetal. Se observaron efectos teratogénicos (onfalocele) en el conejo. Las mujeres en edad fértil deberán emplear métodos anticonceptivos médicamente aceptables durante (y hasta 3 meses después) del período de tratamiento. Como se desconoce el riesgo potencial en el ser humano temsirolimus endovenoso no debe emplearse durante el embarazo. Temsirolimus intravenoso podría ser usado durante el embarazo sólo si los beneficios potenciales justifican los potenciales riesgos para el feto o embrión. Además, los hombres deberán ser debidamente aconsejados antes de comenzar el tratamiento con temsirolimus y es necesario que comprendan el posible riesgo de tomar un medicamento cuyos efectos sobre el feto o esperma aún no se conocen. Los hombres con parejas en edad de procrear deberán utilizar preservativos médicamente aceptables durante todo el tratamiento, recomendándoles continuar con los mismos durante 12 semanas después de la última dosis de temsirolimus. Fertilidad: Estudios en ratas han demostrado una disminución de la fertilidad. Embarazo: No existen datos disponibles sobre el uso de temsirolimus en mujeres embarazadas. No existen estudios adecuados y bien controlados en mujeres embarazadas que consuman temsirolimus. Estudios en animales han demostrado toxicidad reproductiva. Se desconoce el riesgo potencial para los humanos. En estudios sobre la toxicidad animal con ratas y conejos, se observó una mayor mortalidad embrionaria/fetal y un menor crecimiento fetal. Las mujeres en edad reproductiva deben usar contracepción médicamente aceptable durante el tratamiento (y hasta 3 meses después). El temsirolimus por vía intravenosa debe usarse durante el embarazo solamente si el beneficio potencial justifica el riesgo potencial al embrión/feto. Si una paciente queda embarazada durante el tratamiento con temsirolimus, ella y su médico tratante deben analizar exhaustivamente el diagnóstico, las opciones alternativas y los riesgos potenciales del temsirolimus para el feto en desarrollo. Además, los hombres deben recibir asesoramiento adecuado antes de comenzar el tratamiento con temsirolimus y necesitan entender el posible peligro de consumir un medicamento cuyos efectos sobre el feto o esperma se desconocen. Los hombres que están en pareja con mujeres con potencial de reproducción deben usar contracepción médicamente aceptable durante todo el tratamiento y se recomienda que continúen de esa manera durante 12 semanas después de la última dosis de temsirolimus. No existe información disponible para el parto. Período de lactancia: No se han realizado estudios en lactancia del temsirolimus por vía intravenosa. Se desconoce si el temsirolimus se excreta en la leche humana. Debido a que muchos medicamentos se excretan en la leche humana, y debido a que los efectos de la excreción de temsirolimus en la leche humana no se han estudiado, se debe asesorar a las mujeres respecto a la lactancia mientras reciben temsirolimus. Toxicidad:Embarazo: En los estudios de toxicidad sobre el desarrollo con dosis orales en ratas, se observó aumento de la mortalidad embrio/fetal y disminución del desarrollo fetal a dosificaciones >0.45 mg/kg/día (1.1 veces la dosis recomendada en seres humanos ajustada por mg/kg/día y 0.18 veces la dosis recomendada ajustada por mg/m2). En estudios de toxicidad sobre el desarrollo con administración oral en conejos, se observó un incremento en la mortalidad embrional /fetal y disminución del crecimiento fetal a dosis> 0.6 mg /kg /día (1.4 veces la dosis humana recomendada en una base de mg /kg/ día y 0.47 veces la dosis recomendada sobre una base en mg/m2). En conejos se observó una mayor incidencia de protrusión intestinal a través del abdomen con dosis de 0.9 mg/kg/día (2.1 veces la dosis recomendada en seres humanos ajustada por mg/kg/día y 0.7 veces la dosis recomendada en humanos corregida por mg/m2). Carcinogenicidad: No se han realizado estudios de carcinogenicidad con temsirolimus. Mutagenicidad: El temsirolimus no demostró ser genotóxico en una batería de ensayos in vitro (mutación inversa bacteriana en Salmonella typhimurium y Escherichia coli, mutación de avance en células de linfoma de ratones aberraciones cromosómicas en células ováricas de hámster chino) e in vivo (micronúcleos en ratones). Dańo a la fertilidad: En ratas hembra, la fertilidad disminuyó con dosis ≥0.5 mg/kg/día (1.2 veces la dosis recomendada en seres humanos ajustada por mg/kg/día y 0.19 veces la dosis recomendada ajustada por mg/m2). Se observó ausencia de fertilidad a 1 dosificación de 5 mg/kg/día (11.9 veces la dosis recomendada en seres humanos ajustada por mg/kg/día y 1.9 veces la dosis recomendada en seres humanos ajustada por mg/m2). Los efectos sobre la fertilidad en machos estuvieron acompańados por degeneración tubular testicular, disminución de la concentración y motilidad espermática y pesos reducidos de los órganos reproductores con dosificaciones >0.5 mg/kg/día. En ratas hembra, se observó una mayor incidencia de pérdidas pre y post-implantes con dosificaciones ≥0.7 mg/kg/día, que produjo disminución del número de fetos vivos (1.7 veces la dosis recomendada en seres humanos ajustada por mg/kg/día y 0.27 veces la dosis recomendada en seres humanos ajustada por mg/m2). Los pesos fetales disminuyeron con dosificaciones ≥1 mg/kg/día (2.4 veces la dosis recomendada en seres humanos ajustada por mg/kg/día y 0.39 veces la dosis recomendada en seres humanos ajustada por mg/m2).
Interacciones Medicamentosas:Inductores del metabolismo de CYP3A: La coadministración del concentrado inyectable temsirolimus con rifampicina, un potente inductor de CYP3A4/5, no tuvo efectos significativos sobre la Cmax (concentración máxima) y el AUC de temsirolimus después de la administración endovenosa, pero redujo la Cmax de sirolimus en un 65% y AUC por 56%, y la suma del AUC (AUC de Temsirolimus más AUC de sirolimus) en un 41|% en comparación con el tratamiento con temsirolimus solo. Por lo tanto, deberá evitarse el tratamiento concomitante con agentes con potencial de inducción de CYP3A4/5. Si no se pudiera administrar un tratamiento alternativo, deberá considerarse una dosis endovenosa semanal de temsirolimus de hasta 50 mg. Inhibidores del metabolismo de CYP3A: La coadministración de temsirolimus con ketoconazol, un potente inhibidor de CYP3A4, no tuvo efectos significativos sobre la Cmax o el AUC de temsirolimus; sin embargo, el AUC de sirolimus aumentó 3.1 veces y la suma del AUC aumentó 2.3 veces en comparación con temsirolimus solo. Las sustancias que son potentes inhibidores de la actividad de CYP3A4 aumentan las concentraciones sanguíneas del sirolimus. Deberá evitarse el tratamiento concomitante de temsirolimus concentrado inyectable con agentes que tengan fuerte potencial de inhibición de CYP3A4. El tratamiento concomitante con inhibidores moderados de CYP3A4 debe ser únicamente administrado con precaución en pacientes tratados con 25 mg y no debe administrarse a pacientes que reciban dosis superiores a 25 mg de temsirolimus. Interacciones con drogas metabolizadas por CYP2D6: En 23 sujetos sanos la concentración de desipramina, un sustrato de CYP2D6, no se vio alterada cuando se coadministró 25 mg de temsirolimus. No cabe esperar efectos clínicamente significativos cuando se coadministre temsirolimus con agentes metabolizados por CYP2D6. Interacciones con drogas que son sustratos de la P-glucoproteína: En un estudio in vitro, temsirolimus inhibió el transporte de la digoxina, un sustrato de la P-gp, con un valor de CI50 de 2µM. Se desconocen las implicancias clínicas relacionadas con la administración concomitante de sustratos de P-gp.
Sobredosificación: No existe un tratamiento específico para la sobredosis de temsirolimus inyectable; sin embargo, se han administrado dosis endovenosas repetidas de Temsirolimus tan altas como 220 mg/m2 a pacientes con cáncer en forma segura.
Conservación: Temsirolimus Concentrado Inyectable debe ser almacenado bajo refrigeración a 2-8şC y protegido de la luz.
Observaciones: Ver información completa para prescribir en documento de monografía entregado por Pfizer Chile S.A.
 Laboratorio
Laboratorio