Composición:Xalkori 200 mg: Cada cápsula contiene: Crizotinib 200 mg. Xalkori 250 mg: Cada cápsula contiene: Crizotinib 250 mg. Excipientes: Dióxido de Silicio Coloidal, Celulosa Microcristalina, Fosfato de Calcio Dibásico Anhidro, Almidón Glicolato de Sodio, Estearato de Magnesio y cubierta de cápsula de Gelatina dura. Entre los componentes de la cubierta de cápsula de color rosa opaco se incluyen: Gelatina, Dióxido de Titanio y Oxido de Hierro Rojo. Entre los componentes de la cubierta de cápsula de color blanco opaco se incluyen: Gelatina y Dióxido de Titanio. La tinta de impresión contiene Goma Shellac, Propilenglicol, Solución de Amoníaco fuerte, Hidróxido de Potasio y Oxido de Hierro Negro.
Indicaciones: Crizotinib está indicado para el tratamiento del cáncer de pulmón de células no pequeńas (NSCLC) avanzado y positivo a la kinasa de linfoma anaplásico (ALK) previamente tratado. Posología y administración: Pruebas de ALK. La detección del NSCLC positivo a la ALK es necesaria para seleccionar a los pacientes para el tratamiento con crizotinib, ya que este tipo de pacientes son los únicos en los cuales se han demostrado los beneficios.
Posología: La dosis recomendada de crizotinib es de 250 mg por vía oral 2 veces al día. El tratamiento continúa mientras que el paciente obtenga beneficios clínicos de la terapia. Crizotinib puede tomarse acompańado o no de alimentos. Las cápsulas deben tragarse enteras. Si el paciente pasa por alto una de las dosis, debe tomarse apenas lo recuerde, a no ser que falten menos de 6 horas para la siguiente dosis, en cuyo caso el paciente no debe tomar la dosis olvidada. Los pacientes no deben tomar 2 dosis al mismo tiempo para compensar la dosis olvidada. Modificación de la dosis: Puede requerirse la interrupción o la reducción de la dosis según la seguridad y la tolerancia del individuo. Si resulta necesario reducir la dosis, la dosis de crizotinib debe reducirse a 200 mg tomados vía oral 2 veces al día y, si se precisa reducirla aún más, debe reducirse a 250 mg vía oral 1 vez al día. Se proporcionan las pautas para la reducción de dosis para las toxicidades hematológicas y no hematológicas en Tabla 1 y Tabla 2. Tabla 1. Modificación de la dosis de crizotinib: Toxicidades hematológicasa. Grado según los CTCAEb: Grado 3: Dosis de crizotinib: Suspender hasta la recuperación a un grado ≤2, luego reanudar la misma dosis. Grado 4: Suspender hasta la recuperación a un grado ≤2, luego reanudar con 200 mg 2 veces al díac. a Excepto la linfocitopenia (a menos que se relacione con casos clínicos; por ejemplo, infecciones oportunistas). b Criterios comunes terminológicos para la evaluación de las reacciones adversas del NCI.c En caso de recurrencia, suspender hasta la recuperación a un grado ≤2, luego reanudar con 250 mg diarios. Discontinuar su tratamiento de manera permanente en el caso de una recurrencia de Grado 4. Tabla 2. Modificación de la dosis de crizotinib: Toxicidades no hematológicas: Grado según los CTCAEa: Elevación Grado 3 ó 4 de la alanina aminotransferasa (ALT) o de la aspartato aminotransferasa (AST) con elevación de bilirrubina total de Grado <a 1. Dosis de crizotinib: Suspender hasta la recuperación a un Grado ≤1 o a los valores de referencia; luego, reanudar con 200 mg 2 veces al díab. Grado según los CTCAEa: Elevación Grado 2, 3 o 4 de ALT o
la AST con elevación concurrente Grado 2, 3 o 4 de bilirrubina total (en ausencia de colestasis o hemólisis). Dosis de crizotinib: Discontinuar de manera permanente. Grado según los CTCAEa: Neumonitis/enfermedad pulmonar intersticial de cualquier Gradoc. Dosis de crizotinib: Discontinuar de manera permanente. Grado según los CTCAEa: Prolongación de intervalo QTc de Grado 3. Dosis de crizotinib: Suspender hasta la recuperación a un Grado ≤ 1, luego reanudar con 200 mg 2 veces al díab. Grado según los CTCAEa: Prolongación de intervalo QTc de Grado 4. Dosis de crizotinib: Discontinuar de manera permanente.a Criterios comunes terminológicos para la evaluación de las reacciones adversas según el del NCI.b En caso de recurrencia, suspender hasta la recuperación a un Grado ≤ 1, luego reanudar con 250 mg diarios. Discontinuar su tratamiento de manera permanente en el caso de una recurrencia de Grado 3ó 4.c No atribuible a la progresión del cáncer NSCLC, otras enfermedades pulmonares, infecciones o efectos de la radiación. Insuficiencia hepática: Como el crizotinib se metaboliza de manera extensa en el hígado, es probable que la insuficiencia hepática aumente las concentraciones plasmáticas de crizotinib. Sin embargo, no se ha estudiado el crizotinib en pacientes con insuficiencia hepática. Estudios clínicos realizados excluyeron pacientes con ALT o AST >2.5 x LSN o, si se debe a tumores malignos subyacentes, >5.0 x LSN o con bilirrubina total >1.5 x LSN. El tratamiento con crizotinib debe realizarse con precaución en pacientes con insuficiencia hepática leve a moderada (véase la Tabla 2 y la Sección Advertencias y precauciones) No debe administrarse en pacientes con insuficiencia hepática grave. Insuficiencia renal: No se precisa ajustar la dosis inicial en los pacientes con insuficiencia renal leve (depuración de creatinina [CLcr] 60 a 90 ml/min) y moderada (CLcr 30 a 60ml/min). La necesidad potencial de ajustar la dosis inicial en los pacientes con insuficiencia renal grave no puede determinarse, ya que los datos clínicos y farmacocinéticos se encontraron disponibles para un solo paciente. Además, no se encontraron disponibles datos para pacientes con nefropatía terminal. Nińos: No se han establecido la seguridad y la eficacia de crizotinib. Ancianos: No se observaron diferencias generales en cuanto a la seguridad o a la eficacia en comparación con pacientes más jóvenes.
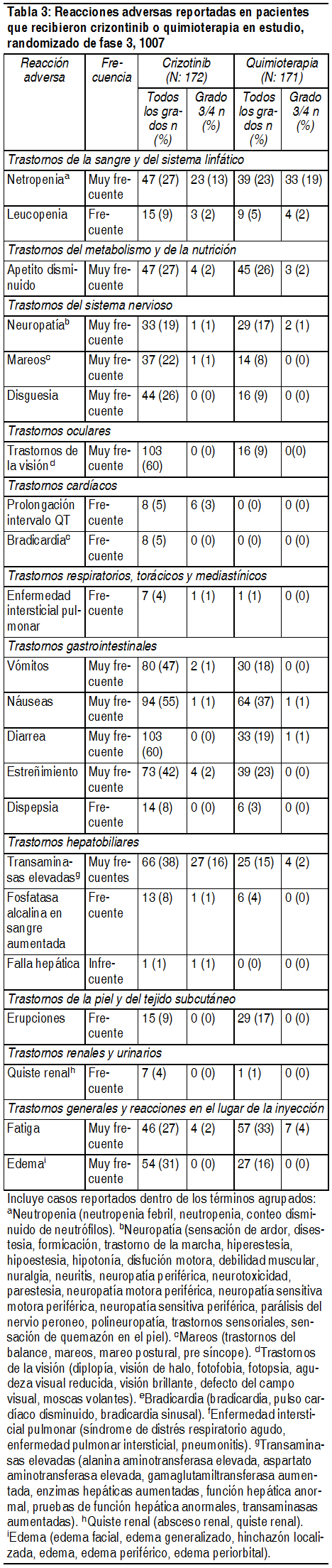
Efectos Colaterales:Resumen de perfil de seguridad: La información descrita a continuación refleja la exposición a crizotinib en 172 pacientes con cáncer de pulmón de células no pequeńas (NSCLC) avanzado y positivo a la kinasa de linfoma anaplásico (ALK) que participaron en el Estudio, randomizado de fase 3, 1007 y en 1053 pacientes con cáncer de pulmón de células no pequeńas (NSCLC) avanzado y positivo a la kinasa de linfoma anaplásico (ALK) que participaron en 2 Estudios clínicos de rama única (Estudios 1001 y 1005). Estudio 1007 randomizado de Fase 3: La población de análisis de seguridad en el Estudio 1007 randomizado de fase 3 incluyó 172 pacientes que recibieron crizotinib y 171 pacientes que recibieron quimioterapia (99 con premetrexed, 72 docetaxel). La duración media del tratamiento en estudio fue de 31 semanas para pacientes recibiendo crizotinib y 12 semanas para pacientes con quimioterapia. Interrupciones de dosis debidas a reacciones adversas relacionadas al tratamiento ocurrieron en 54 (31%) pacientes recibiendo crizotinib y en 14 (8%) pacientes con quimioterapia. Reducciones adversas debidas a reacciones adversas relacionadas al tratamiento ocurrieron en 26 (15%) pacientes recibiendo crizotinib y en 24 (14%) recibiendo quimioterapia. Efectos adversos relacionados al tratamiento resultantes en discontinuación permanente ocurrieron en 11 (6%) pacientes recibiendo crizotinib y en 17 (10%) recibiendo quimioterapia. La Tabla 3 compara las reacciones adversas experimentadas por pacientes en las ramas de crizotinib y quimioterapia del estudio randomizado de fase 3 1007. Nota: Las reacciones adversas están listadas por clase de sistema de órganos, categoría de frecuencia y grado de severidad. Las frecuencias basadas en la incidencia de crizotinib se definen como: muy frecuente (≥ 1/10), frecuente ≥ 1/100 a < 1/10), infrecuente (≥ 1/1000 a < 1/100), raras (≥ 1/10,000 a < 1/1000), muy raras (<1/10000).
Estudios de rama única en NSCLC avanzado ALK-positivo: La población de análisis de seguridad en el Estudio 1005 incluyó 934 pacientes que recibieron crizotinib. La duración media del tratamiento fue de 23 semanas. Las interrupciones y reducciones de dosis debido a efectos adversos relacionados al tratamiento ocurrieron en 212 (23%) pacientes y en 116 (12%) pacientes en el Estudio 1005, respectivamente. Efectos adversos relacionados al tratamiento resultantes en discontinuación permanente ocurrieron en 45 (5%) pacientes en el Estudio 1005. Los efectos adversos relacionados al tratamiento más comunes (≥25%) en el estudio 1005 incluyeron trastornos de la visión, náuseas, vómitos, diarrea, edema, estreńimiento y fatiga. Los efectos adversos más comunes de Grado 3 ó 4 relacionados al tratamiento (≥3%) en el estudio 1005 fueron neutropenia, transaminasas elevadas, y estreńimiento. La población de análisis de seguridad en el Estudio 1001 incluyó 119 pacientes que recibieron crizotinib. La duración media del tratamiento fue de 32 semanas. Las interrupciones y reducciones de dosis debido a efectos adversos relacionados al tratamiento ocurrieron en 14 (12%) pacientes y en 5 (4%) pacientes en el Estudio 1001, respectivamente. Efectos adversos relacionados al tratamiento resultantes en discontinuación permanente ocurrieron en 3 (3%) pacientes en el Estudio 1001. Los efectos adversos relacionados al tratamiento más comunes (≥25%) en el estudio 1001fueron consistentes con los del estudio, randomizado de fase 3, 1007 y con los del estudio de rama única 1005, e incluyen trastornos de la visión, náuseas, vómitos, diarrea, edema, estreńimiento, fatiga y mareos. Efectos visuales: Entre los trastornos trastornos de la visión de distintas causas producto del tratamiento, los impedimentos visuales más frecuente como, fotopsia, visión borrosa, moscas volantes, se experimentaron por 103 (60%) pacientes en el Estudio randomizado de fase 3 1007, y 75 (63%) y 513 (55%) pacientes en los Estudios 1001, y 1005 respectivamente. Trastornos de la visión relacionados al tratamiento evaluados por investigadores, fueron reportados por 101 (59%) pacientes en el estudio randomizado de fase 3 1007, y 74 (62%) y 496 (53%) pacientes en los Estudios 1001 y 1005, respectivamente. Más de 96% de estos pacientes tuvieron episodios de severidad leve con tiempos medios de inicio de 5 días, 13 días, y 7 días en los Estudios 1007, 1001, y 1005, respectivamente. Se debe considerar una evaluación oftalmológica si persiste el trastorno de la visión o si este empeora. Cuatro pacientes precisaron discontinuación temporal del tratamiento y 1 paciente precisó reducción de dosis por trastornos visuales, todos del estudio 1005. No se requirió la discontinuación permanente del tratamiento con crizotinib debido a trastornos visuales para ningún paciente en los estudios 1007,1001 y 1005. Efectos gastrointestinales: Náuseas, diarrea, vómitos y estreńimiento fueron los casos gastrointestinales más informados. Los tiempos medios de inicio para nauseas y vómitos fue de 2 a 3 días. La mayoría de los episodios fueron de severidad leve a moderada, y disminuyeron en su frecuencia después de 3 a 4 semanas de tratamiento. El tratamiento de soporte debiese incluir el uso de medicamentos antieméticos. En estudios clínicos, los medicamentos eméticos más comúnmente usados fueron ondansetron y proclorperazina. Diarrea y constipación fueron principalmente leves a moderados en severidad. El tratamiento de soporte para diarrea y constipación debiese incluir el uso de antidiarreicos estándar y medicamentos laxantes, respectivamente. Efectos del sistema nervioso: Neuropatía producto del tratamiento de distintas causas, como se define en la Tabla 3, principalmente neuropatía periférica, fue experimentada por 33(19%) pacientes en el Estudio randomizado de fase 3 1007, y 24 (20%) y 178 (19%) pacientes en los estudios 1001 y 1005, respectivamente. Neuropatía relacionada al tratamiento evaluada por investigadores fue experimentada por 14 (8%) pacientes en el Estudio randomizado de fase 3 1007 y 13 (11%) y 95 (10%) pacientes en los Estudios 1001 y 1005, respectivamente, y fue primordialmente de gravedad Grado 1. También se informaron con frecuencia mareos y disgeusia en estos estudios, pero todos fueron de gravedad Grado 1. Bradicardia: Se experimentó bradicardia por distintas causas producto del tratamiento por 8 (5%) pacientes en el Estudio, randomizado de fase 3, 1007 y por 8 (7%) y 57 (6%) pacientes en los Estudios 1001 y 1005, respectivamente. La bradicardia evaluada por investigadores relacionada al tratamiento se experimentó por 7 (4%) pacientes en el Estudio, randomizado de fase 3, 1007 y por 6 (5%) y 49 (5%) pacientes en los Estudios 1001 y 1005, respectivamente. La mayoría de estos casos fueron de severidad Grado 1 ó 2. En los Estudios 1007, 1001 y 1005, 19 de 170 (11%) pacientes, 16 de 114 (14%) pacientes, y 90 de 890 (10%) pacientes presentaron frecuencia de pulso <50 lpm. El uso de medicamentos concomitantes asociados a bradicardia debe ser cuidadosamente evaluado. Los pacientes que desarrollan bradicardia sintomática deben manejarse como se recomienda en las secciones de modificación de dosis y advertencias y precauciones. Quiste renal: Quistes renales complejos por distintas causas producto del tratamiento fueron experimentados por 7 (4%) pacientes en el Estudio randomizado de fase 3 1007 y 0 y 12 (1%) pacientes en los Estudios 1001 y 1005, respectivamente. Los quistes renales complejos relacionados al tratamiento evaluados por investigadores se reportaron en 7 (4%) pacientes en el Estudio 1007 randomizado de fase 3 y 11 (1%) pacientes en el Estudio 1005. No hubo reportes de análisis de orina anormales o de insuficiencia renal clínicamente relevantes en estos casos, aunque en algunos pacientes se observó invasión quística local más allá del rińón. En pacientes que desarrollan quistes renales se deben considerar monitoreos periódicos de imágenes y análisis de orina.
Contraindicaciones: Crizotinib está contraindicado en pacientes con hipersensibilidad a crizotinib o a cualquiera de sus excipientes.
Advertencias:Hepatotoxicidad: Han ocurrido casos de hepatotoxicidad inducida por la medicación con resultados mortales. Estos casos ocurrieron durante el tratamiento con crizotinib en menos del 1% de los pacientes en estudios clínicos. Las elevaciones simultáneas en la ALT mayor a 3 x LSN y la bilirrubina total mayor a 2 x LSN sin la fosfatasa alcalina elevada se han observado en menos del 1% de los pacientes en estudios clínicos. Las elevaciones de Grado 3 y 4 fueron generalmente asintomáticas y reversibles al momento de la interrupción del tratamiento. Por lo general, los pacientes reanudaron el tratamiento en dosis menores sin recaídas; sin embargo, 1 paciente del Estudio A (<1%) y 3 pacientes del Estudio B (2%) 2 pacientes del Estudio randomizado de fase 3 1007 (1%), 1 paciente del Estudio 1001 (<1%), y 6 pacientes del estudio 1005 (<1%) precisaron la interrupción permanente del tratamiento. Las elevaciones de aminotransferasa ocurrieron generalmente dentro de los primeros 2 meses del tratamiento. Las pruebas de función hepática, incluidas la ALT, AST y la bilirrubina total, deben controlarse 1 vez por mes y como se indica clínicamente, con repeticiones de pruebas más frecuentes para las elevaciones de los Grados 2, 3 ó 4. Para los pacientes que desarrollan aumento de transaminasas, véase la sección Modificación de la dosis. Enfermedad pulmonar intersticial (neumonitis): Crizotinib se ha relacionado con enfermedad pulmonar intersticial (EPI)/neumonitis grave, potencialmente mortal o mortal bajo tratamiento en estudios clínicos. Se debe controlar la aparición de síntomas que indiquen enfermedad pulmonar intersticial (EPI)/neumonitis. Deben excluirse otras causas de la enfermedad pulmonar intersticial (EPI)/neumonitis Los pacientes deben discontinuar de manera permanente el tratamiento con crizotinib si se les diagnostica enfermedad pulmonar intersticial (EPI)/neumonitis por el tratamiento. Prolongación del intervalo QT: Se ha observado la prolongación del intervalo QTc sin arritmia. Crizotinib debe administrarse con precaución a los pacientes con antecedentes de o con predisposición para la prolongación del intervalo QTc, o a quienes toman medicación que se conoce por prolongar el intervalo QT. Cuando este tipo de pacientes toman crizotinib, se debe considerar la realización de controles periódicos con electrocardiogramas y electrólitos. Para los pacientes con prolongación del intervalo QTc, véase la sección Modificación de la dosis. Bradicardia: Se ha reportado bradicardia en estudios clínicos, generalmente asintomática. El efecto total de crizotinib en la frecuencia de pulso puede no desarrollarse hasta varias semanas después de iniciado el tratamiento. Se debe evitar el uso de crizotinib en combinación con otros agentes bradicórdicos (ej., beta-bloqueadores, bloqueadores de canal de calcio no dihidropiridínicos como verapamilo y diltiazem, clonidina, digoxina) en el grado posible, debido al riesgo aumentado de bradicardia sintomática (síncope, mareos, hipotensión). Se recomiendan monitoreos mensuales de frecuencia de pulso y de presión sanguínea. No se requiere modificación de dosis en casos de bradicardia asintomática. En casos de bradicardia sintomática, crizotinib debe suspenderse y el uso de medicamentos concomitantes debe reevaluarse. Para el manejo de pacientes que desarrollan bradicardia sintomática, ver las secciones de Modificación de dosis y Efectos colaterales. Insuficiencia renal: Si los pacientes presentan insuficiencia renal severa que no requiere diálisis peritoneal o hemodiálisis, la dosis de crizotinib debe ser ajustada.
Precauciones:Fertilidad: Según resultados de seguridad no clínicos, es probable que la fertilidad tanto en varones como en mujeres se encuentre comprometida por el tratamiento con crizotinib. No se han observado diferencias generales en cuanto a la seguridad o a la eficacia en comparación con pacientes más jóvenes. Embarazo: Crizotinib puede ocasionar dańo fetal si se la administra a una mujer embarazada. No se estableció que el crizotinib sea teratogénico en ratas o conejas preńadas. Entre los efectos adversos, se consideró la reducción en el peso corporal de los fetos de ratas y conejos a 200 y 60 mg/kg/día, respectivamente (aproximadamente 2 veces la exposición clínica humana según la AUC).No existen estudios adecuados y bien controlados en mujeres embarazadas que tomancrizotinib. Las mujeres con probabilidades de embarazo deben ser advertidas para evitar el embarazo durante el tratamiento con crizotinib. Cuando tomen este medicamento, estas mujeres o sus parejas deben usar métodos anticonceptivos adecuados durante el tratamiento y por al menos 90 días posteriores al haberlo finalizado. Las mujeres que tomen crizotinib durante el embarazo o que queden embarazadas durante el tratamiento deben ser advertidas sobre los riesgos que corre el feto. Los varones que tomen crizotinib también deben ser advertidos de esto si su pareja se encuentra embarazada o con probabilidades de estarlo. Lactancia: Se desconoce si crizotinib o sus metabolitos se excretan en la leche humana. Debido a que muchos medicamentos se excretan en la leche humana, y debido al riesgo de la aparición de reacciones adversas graves durante el período de lactancia por la exposición al crizotinib, se debe elegir entre discontinuar la lactancia o la toma del medicamento, tomando en cuenta la importancia de este para la madre. Uso pediátrico: No se han establecido la seguridad y eficacia de crizotinib. Uso geriátrico: No se han observado diferencias generales en cuanto a la seguridad o a la eficacia en comparación con pacientes más jóvenes. Efectos sobre las actividades que requieren concentración y desempeńo: No se han realizado estudios con respecto a los efectos de crizotinib sobre la capacidad de conducir y de usar máquinas. Sin embargo, se deben tomar precauciones al conducir o al operar maquinarias, sobre todo por parte de los pacientes con trastornos en la visión, mareos o cansancio durante el tratamiento con crizotinib.
Interacciones Medicamentosas:Agentes que pueden aumentar las concentraciones plasmáticas: La administración conjunta de crizotinib con potentes inhibidores de CYP3A puede incrementar las concentraciones plasmáticas de crizotinib. Debe evitarse el uso concomitante de inhibidores de CYP3A, incluidos, entre otros: atazanavir, claritromicina, indinavir, itraconazol, ketoconazol, nefazodona, nelfinavir, ritonavir, saquinavir, telitromicina, troleandomicina y voriconazol. La toronja (o pomelo) o su jugo también pueden incrementar las concentraciones plasmáticas de crizotinib y debe evitarse. Agentes que pueden disminuir las concentraciones plasmáticas: La administración conjunta de crizotinib con potentes inductores de CYP3A puede disminuir las concentraciones plasmáticas de crizotinib. Debe evitarse el uso simultáneo de inductores de CYP3A, incluidos, entre otros: carbamazepina, fenobarbital, fenitoína, rifabutina, rifampina y hierba de San Juan o hipérico. Agentes cuyas concentraciones plasmáticas pueden alterarse por crizotinib: Crizotinib se ha identificado como inhibidor de CYP3A tanto in vitro como in vivo. Debe tenerse precaución si se administra crizotinib junto con medicamentos que se metabolizan principalmente por el CYP3A, en particular aquellos sustratos de CYP3A con estrechos índices terapéuticos, incluidos, entre otros: alfentanilo, ciclosporina, fentanilo, quinidina, sirolimus y tacrolimus. Debe evitarse la administración conjunta de crizotinib con los sustratos de CYP3A con estrechos índices terapéuticos y relacionados con arritmias potencialmente mortales, incluidos, entre otros: dihidroergotamina, ergotamina y pimozida. Nota a los países: Aunque no se comercializa en todos los países, los sustratos de CYP3Aastemizola, cisaprida y terfenadina deben evitarse, ya que también tienen estrechos índices terapéuticos y se han relacionado con arritmias potencialmente mortales. Interacciones con pruebas de laboratorio y otras pruebas de diagnóstico: Anormalidades hematológicas de laboratorio. En el Estudio randomizado de fase 3 1007, se observaron cambios de Grado 3 ó 4 de reducción de leucocitos y neutrófilos en frecuencias de 5% y 13%, respectivamente. En el Estudio 1001, se observaron cambios de Grado 3 ó 4 de reducción de tanto leucocitos como de neutrófilos en frecuencias de <4%. En el estudio 1005 se observaron cambios de Grado 3 ó 4 de reducción de leucocitos y neutrófilos en frecuencias de <3% y 8%, respectivamente. Debe realizarse un control de hemograma completo, incluidos los recuentos y fórmulas leucocitarias como se indica clínicamente, con análisis más frecuentes si se observan anormalidades de Grados 3 ó 4, o si se presenta fiebre o infección. En los pacientes que desarrollan anormalidades hematológicas de laboratorio, véase la sección modificación de la dosis. Anormalidades hepáticas de Laboratorio. En el Estudio randomizado de fase 3 1007, aumentos a Grado 3 ó 4 de ALT, AST, y fosfatasa alacalina se observaron en los pacientes en frecuencias de 17%, 9% y 2% respectivamente. En el Estudio 1001, aumentos a Grado 3 ó 4 de ALT, AST, y fosfatasa alacalina se observaron en los pacientes en frecuencias de 5%, 3% y 0% respectivamente. En el Estudio 1005, aumentos a Grado 3 ó 4 de ALT, AST, y fosfatasa alacalina se observaron en los pacientes en frecuencias de 8%, 4% y 2% respectivamente. Los pacientes deben ser monitoreados por hepatotoxicidad y manejados según lo recomendado en la sección de Advertencias y Precauciones.
Sobredosificación: No se han presentado casos de sobredosis de crizotinib. El tratamiento por sobredosis con crizotinib debe incluir medidas de apoyo generales. No hay antídoto para crizotinib.
 Laboratorio
Laboratorio