Composición: Cada comprimido recubierto contiene: 50 mg de Dolutegravir. Los comprimidos son de color azul verdoso, con forma de cápsula y grabados en una cara con GX LL1. Lista de excipientes: Núcleo del comprimido: D-Manitol; Celulosa Microcristalina; Povidona K29/32; Almidón Glicolato de Sodio; Estearil Fumarato de Sodio. Cubierta del comprimido: Alcohol Polivinílico - Parcialmente Hidrolizado; Dióxido de Titanio; Macrogol/PEG; Talco; Óxido de Hierro Amarillo.
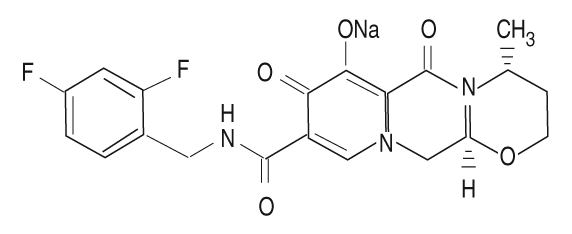
Descripción: Tivicay contiene dolutegravir, como dolutegravir sódico, un inhibidor de la integrasa del HIV. El nombre químico de dolutegravir sódico es: sodio (4R, 12aS) -9 - {[(2.4 difluorofenil) metil] carbamoil} - 4-metil-6, 8-dioxo-3, 4, 6, 8,12, 12a -hexahidro-2H-pirido [1 ', 2': 4,5] pirazino [2.1-b] [1.3] oxazin-7-olato. La fórmula empírica es C20H18F2N3NaO5 y el peso molecular es 441.36 g/mol.
Tiene la siguiente fórmula estructural:
Dolutegravir sódico es un polvo entre color blanco y amarillo claro y es ligeramente soluble en agua.
Indicaciones: Tivicay está indicado en combinación con otros agentes antirretrovirales para el tratamiento de la infección por el virus de la inmunodeficiencia humana tipo 1 (HIV-1). Limitaciones de uso: El uso de Tivicay en pacientes experimentados con inhibidores de la transferencia de cadenas de integrasa (INSTI) debe estar guiado por el número y tipo de sustituciones INSTI basales. La eficacia de Tivicay 50 mg administrado 2 veces al día, se reduce en pacientes con una sustitución de resistencia INSTI Q148 más 2 o más sustituciones de resistencia INSTI adicionales incluyendo T66A, L74I/M, E138A/K/T, G140S/A/C, Y143R/C/H, E157Q, G163S/E/K/Q, o G193E/R (véase Microbiología).
Propiedades:Farmacología clínica: Mecanismo de acción: Dolutegravir es un agente antiviral HIV-1(véase Microbiología). Farmacodinamia: En un estudio aleatorizado, de hallazgo de dosis, los sujetos infectados por el HIV -1 tratados con monoterapia con dolutegravir demostraron actividad antiviral rápida y dependiente de la dosis con caídas medias desde el inicio hasta el día 11 en el ARN del HIV-1 de 1.5; 2.0 y 2.5 log10 para dolutegravir 2 mg, 10 mg y 50 mg 1 vez al día, respectivamente. Esta respuesta antiviral se mantuvo durante los 3 a 4 días después de la última dosis en el brazo de 50 mg. Efectos sobre el electrocardiograma: En un ensayo aleatorizado, cruzado, controlado con placebo, 42 sujetos sanos recibieron una dosis oral de placebo. Dolutegravir 250 mg suspensión (exposiciones aproximadamente 3 veces más de la dosis de 50 mg 1 vez al día en estado estacionario), y moxifloxacino 400 mg (control activo) de forma aleatoria. Después del ajuste basal y para placebo, la media máxima del cambio en el QTc basado en el método de corrección Fridericia (QTcF) para dolutegravir fue de 2.4 mseg (CI superior unilateral del 95%: 4.9 mseg). Tivicay no prolongó el intervalo QTc durante las 24 horas después de la dosis. Efectos sobre la función renal: Se evaluó el efecto de dolutegravir sobre la función renal en un ensayo abierto, aleatorio, de 3 brazos, paralelo, controlado con placebo en sujetos sanos (n = 37) que recibieron dolutegravir 50 mg 1 vez al día (n = 12), dolutegravir 50 mg 2 veces al día (n = 13), o placebo 1 vez al día (n = 12) durante 14 días. Una disminución en la depuración de creatinina, determinada por la recolección de orina de 24 horas, se observó con ambas dosis de dolutegravir después de 14 días de tratamiento en los sujetos que recibieron 50 mg 1 vez al día (9% de disminución) y 50 mg 2 veces al día (13% de disminución). Ninguna de las dosis de dolutegravir tuvo un efecto significativo en la tasa de filtración glomerular real (determinado por la eliminación del sustrato de prueba, iohexol) o el flujo plasmático renal efectivo (determinado por la eliminación del sustrato de prueba, para-amino hipurato) en comparación con el placebo. Farmacocinética: Las propiedades farmacocinéticas de dolutegravir se han evaluado en sujetos adultos sanos y en sujetos adultos infectados por HIV-1. La exposición a dolutegravir fue similar entre sujetos sanos y sujetos infectados por HIV-1. La exposición no lineal de dolutegravir después de 50 mg 2 veces al día en comparación con 50 mg 1 vez al día en pacientes infectados por HIV-1 (Tabla 6) se atribuyó al uso de inductores metabólicos en los tratamientos de fondo antirretrovirales de los sujetos que recibieron dolutegravir 50 mg 2 veces al día en los ensayos clínicos. En estos ensayos, Tivicay se administró sin tener en cuenta la alimentación.
Absorción: Tras la administración oral de dolutegravir, las concentraciones máximas se observaron 2 a 3 horas después de la administración. Con administración 1 vez al día, el estado estacionario farmacocinético se alcanza en aproximadamente 5 días, con tasas promedio de acumulación de AUC, Cmax y C24h oscilando entre 1.2 y 1.5 días. Las concentraciones plasmáticas de dolutegravir aumentaron en menor grado a la dosis proporcional sobre 50 mg. Dolutegravir es un sustrato de la glicoproteína-P in vitro. La biodisponibilidad absoluta de dolutegravir no se ha establecido. Efecto de los alimentos sobre la absorción oral: Tivicay puede administrarse con o sin alimentos. Los alimentos aumentaron el grado de absorción y redujeron la velocidad de absorción de dolutegravir. Las comidas bajas, moderadas, y altas en grasa aumentaron el AUC de dolutegravir (0 -∞) en un 33%, 41% y 66%; aumentaron la Cmáx en un 46%, 52% y 67%, y prolongaron el Tmáx a 3, 4, y 5 horas en comparación con 2 horas bajo condiciones de ayuno, respectivamente. Distribución: Dolutegravir presenta una alta unión a proteínas plasmáticas humanas (≥98.9%), en base a los datos in vivo y la unión es independiente de la concentración plasmática de dolutegravir. El volumen de distribución aparente (Vd/ F) tras la administración de una dosis diaria de 50 mg se estima en 17.4 l basado en un análisis farmacocinético de la población. El líquido cefalorraquídeo (LCR): En 11 sujetos sin tratamiento previo con un régimen de dolutegravir 50 mg al día más abacavir/lamivudina, la concentración de dolutegravir en el LCR promedió 18 ng/ml (rango: 4 ng/ml a 23.2 ng/ml) 2 a 6 horas post-dosis después de 2 semanas de tratamiento. La relevancia clínica de este hallazgo no ha sido establecida. Metabolismo y eliminación: Dolutegravir se metaboliza principalmente a través del UGT1A1, con alguna contribución de CYP3A. Después de una sola dosis oral de dolutegravir, el 53% de la dosis oral total se excreta sin cambios en las heces. Treinta y uno por ciento de la dosis oral total se excreta en la orina, representada por el éter glucurónido de DTG (18.9% de la dosis total), por el metabolito formado por la oxidación en el carbón bencílico (3.0% de la dosis total), y por el metabolito de N-dealquilación hidrolítica (3.6% de la dosis total). La eliminación renal del fármaco inalterado fue baja (< 1% de la dosis). Dolutegravir tiene una vida media terminal de aproximadamente 14 horas y una eliminación aparente (CL/F) de 1.0 l/h sobre la base de los análisis farmacocinéticos de la población. Polimorfismos en las enzimas metabolizantes de fármacos: En un metaanálisis utilizando muestras de farmacogenómica recolectadas en los estudios clínicos de sujetos sanos, los sujetos con genotipos UGT1A1 (n= 7) que confieren un pobre metabolismo de dolutegravir, mostraron una eliminación 32% menor de dolutegravir, y 46% mostraron AUC más altas en comparación con sujetos con genotipos asociados con un metabolismo UGT1A1 normal (n= 41). Poblaciones especiales: Insuficiencia hepática: Dolutegravir se metaboliza y es eliminado principalmente por el hígado. En un ensayo que comparaba 8 sujetos con insuficiencia hepática moderada (puntuación de Child-Pugh B) con 8 controles sanos, la exposición a dolutegravir a dosis única de 50 mg fue similar entre los 2 brazos. Un ajuste de dosis no es necesario para pacientes con insuficiencia hepática leve a moderada (puntuación de Child-Pugh A o B). El efecto de la insuficiencia hepática severa (puntuación de Child-Pugh C) sobre la farmacocinética de dolutegravir no se ha estudiado. Por lo tanto, Tivicay no está recomendado para uso en pacientes con insuficiencia hepática severa. Coinfección con VHB/VHC: El análisis farmacocinético poblacional utilizando datos agrupados, no indicó ningún efecto clínicamente relevante de la coinfección con VHC en la farmacocinética de dolutegravir. Hubo información limitada sobre la coinfección con VHB. Insuficiencia renal: La eliminación renal del fármaco inalterado es una vía menor de eliminación de dolutegravir. En un estudio que comparaba 8 sujetos con insuficiencia renal severa (depuración de creatinina <30 ml/min) con 8 controles pareados sanos, las AUC, Cmáx y C24 de dolutegravir se redujeron en un 40%, 23% y 43%, respectivamente, en comparación con los sujetos pareados sanos. La causa de esta disminución se desconoce. El análisis farmacocinético poblacional utilizando datos de los ensayos SAILING y VIKING-3 indicó que la insuficiencia renal leve y moderada no tuvo ningún efecto clínicamente relevante en la exposición de dolutegravir. No es necesario ajustar la dosis para pacientes sin tratamiento previo o con tratamiento previo pero sin inhibidores de la integrasa (INI naďve) con insuficiencia renal leve, moderada o severa o para pacientes con tratamiento INI previo (con ciertas sustituciones de resistencia asociadas con INI o sospecha clínica de resistencia a los INI) con insuficiencia renal leve o moderada. Se debe tener precaución en pacientes con tratamiento con INI previo (con ciertas sustituciones de resistencia INI o sospecha clínica de resistencia a INI (véase Microbiología) con insuficiencia renal severa, ya que la disminución de las concentraciones de dolutegravir puede resultar en pérdida del efecto terapéutico y el desarrollo de resistencia a Tivicay u otros antirretrovirales coadministrados. Dolutegravir no se ha estudiado en pacientes que requieren diálisis. Género: Los análisis farmacocinéticos poblacionales con datos agrupados de ensayos en adultos indican que el género no tuvo ningún efecto clínicamente relevante en la exposición de dolutegravir. Raza: Los análisis farmacocinéticos poblacionales con datos agrupados de ensayos en adultos, indican que la raza de la persona no tuvo ningún efecto clínicamente relevante sobre la farmacocinética de dolutegravir. Pacientes geriátricos: Los análisis farmacocinéticos poblacionales con datos agrupados de ensayos en adultos indican que la edad no tuvo ningún efecto clínicamente relevante sobre la farmacocinética de dolutegravir. Pacientes pediátricos: La farmacocinética de dolutegravir en nińos infectados con HIV-1 (n = 10) mayores o igual a 12 ańos y menores de 18 ańos fue similar a la observada en adultos infectados con HIV-1 que recibieron dolutegravir 50 mg una vez al día (Tabla 7) [véase Estudios clínicos].
Interacciones farmacológicas: Se realizaron ensayos de interacción farmacológica con Tivicay y otras drogas que podrían ser coadministradas o utilizadas como pruebas para las interacciones farmacocinéticas. Como no se espera que dolutegravir afecte la farmacocinética de otros fármacos que dependen del metabolismo hepático (Tabla 8) (véase Interacciones farmacológicas) el objetivo principal de estos ensayos de interacción farmacológica fue evaluar el efecto sobre dolutegravir del fármaco coadministrado (Tabla 9). En la tabla 5 se proporcionan recomendaciones de dosis o régimen, como resultado de las interacciones entre fármacos establecidos y otras interacciones farmacológicas con Tivicay potencialmente significativas (véase Posología e Interacciones farmacológicas).
Microbiología: Mecanismo de acción: Dolutegravir inhibe la integrasa del HIV uniéndose al sitio activo de la integrasa y bloqueando el paso de transferencia de hebra de la integración al ácido desoxirribonucleico (ADN) retroviral, el cual es esencial para el ciclo de replicación del HIV. Ensayos bioquímicos de transferencia de hebra utilizando Integrasa del HIV-1 purificada y ADN sustrato pre-procesado dieron como resultado valores de IC50 de 2.7 nM y 12.6 nM. Actividad antiviral en cultivo celular: Dolutegravir mostró actividad antiviral contra cepas de laboratorio de tipo salvaje HIV-1 con una media de valores de CE50 de 0.5 nM (0.21 ng/ml) a 2.1 nM (0.85 ng/ml) en células mononucleares de sangre periférica (PBMC) y células MT-4. Dolutegravir exhibió actividad antiviral contra 13 aislados clínicos diferentes de clade B con una media de EC50 de 0.52 nM en un ensayo de susceptibilidad de la integrasa viral utilizando la región codificante de la integrasa a partir de aislados clínicos. Dolutegravir demostró actividad antiviral en cultivo celular frente a un panel de aislados clínicos de HIV-1 (3 en cada grupo de M clases A, B, C, D, E, F, y G, y 3 en el grupo O) con valores de EC50 entre 0.02 nM y 2.14 nM para el HIV-1. En los ensayos de PBMC, los valores de EC50 de dolutegravir frente a 3 aislados clínicos de HIV-2 oscilaron entre 0.09 nM y 0.61 nM. Actividad antiviral en combinación con otros agentes antivirales: La actividad antiviral de dolutegravir no fue antagónica al combinarse con el INSTI, raltegravir; los inhibidores no nucleótidos de la transcriptasa reversa (INNTR), efavirenz o nevirapina; los inhibidores nucleósidos/nucleótidos de la transcriptasa reversa (INTR) abacavir o estavudina; los inhibidores de la proteasa (IP), amprenavir o lopinavir ; el antagonista del correceptor CCR5, maraviroc; o el inhibidor de la fusión, enfuvirtida. La actividad antiviral de dolutegravir no fue antagónica al combinarse con el inhibidor de la transcriptasa reversa del VHB, adefovir, o con el antiviral, ribavirina. Resistencia: Cultivo de células: Virus resistentes a dolutegravir fueron seleccionados en cultivo celular a partir de diferentes cepas y clados de tipo salvaje HIV-1. Las sustituciones de aminoácidos E92Q, G118R, S153F o Y, G193E o R263K surgieron en diferentes pasajes y confirieron una susceptibilidad a dolutegravir de hasta 4 veces más reducida. Pasaje de los virus mutantes que contienen las sustituciones Q148R o Q148H seleccionaron para sustituciones adicionales en integrasa que confirieron susceptibilidad reducida a dolutegravir (factor de cambio aumento de 13 a 46). Las sustituciones de integrasa adicionales incluyeron T97A, E138K, G140S y M154I. Pasaje de los virus mutantes que contienen tanto G140S como Q148H seleccionaron para L74M, E92Q y N155H. Sujetos sin tratamiento previo (naďve): Ningún sujeto en los brazos de tratamiento de dolutegravir 50 mg 1 vez al día de los estudios para sujetos sin tratamiento previo (naďve) SPRING-2 y SINGLE experimentaron una disminución detectable en susceptibilidad a dolutegravir o a los INTR de fondo en el subgrupo de análisis de resistencia (n = 6 con el RNA de HIV-1 > 400 copias/ml al momento de fracaso o en la última visita hasta la semana 48 y que tengan datos de resistencia). Un sujeto adicional en SINGLE con 275 copias/ml de ARN de HIV-1 tuvo una sustitución de resistencia-INI (E157Q/P) emergente al tratamiento detectado a la semana 24, pero sin disminución correspondiente en la susceptibilidad a dolutegravir. No se aisló ninguna resistencia genotípica de régimen de fondo emergente al tratamiento en el brazo de dolutegravir en cualquiera de los ensayos SPRING-2 o SINGLE. No se observó ninguna resistencia primaria de sustitución de tratamiento emergente en cualquiera de los grupos de tratamiento del estudio FLAMINGO. Sujetos con tratamiento previo sin uso previo de inhibidores de transferencia de hebra (INI-naďve): En el brazo dolutegravir del estudio SAILING para sujetos con tratamiento previo con antirretrovirales sin INSTI (n = 354), se observaron sustituciones de la integrasa emergentes al tratamiento en 6 de 28 (21%) sujetos que tenían fallas virológicas y antecedentes de resistencia. En 5 de los 6 sujetos aislados se incluyeron sustituciones emergentes a INSTI en L74L/M/I, Q95Q/L, V151V/I (n = 1 cada uno) y R263K (n = 2). El cambio en la susceptibilidad fenotípica de dolutegravir para estos 5 sujetos aislados fue 2 veces menor. Un sujeto aislado presentaba resistencia preexistente a sustituciones de raltegravir E138A, G140S, y Q148H a nivel basal y tenía adicionalmente resistencia a sustituciones emergentes de INSTI T97A y E138A/T con la correspondiente reducción de 148 veces en la susceptibilidad a fallo en dolutegravir. En el brazo comparador de raltegravir, 21 de 49 sujetos (43%) con datos de resistencia post-basal tenían evidencia de sustituciones de resistencia INSTI emergentes (L74M, E92Q, T97A, G140S/A, Y143R/C, Q148H/R, V151I, N155H, E157Q, y G163K/R) y resistencia fenotípica a raltegravir. Sujetos con tratamiento previo con inhibidores de la transferencia de hebra (INSTI): VIKING-3 examinó la eficacia de dolutegravir 50 mg dos veces al día junto a un régimen de base optimizado en pacientes con fracaso virológico previo o actual de regímenes que contenían inhibidores de la integrasa INSTI (raltegravir o elvitegravir). Respuesta en base a genotipo basal: De los 183 sujetos con datos basales, 30% albergaron el virus con una sustitución en Q148, y 33% no tenían sustituciones primarias a la resistencia INSTI (T66A/I/K, E92Q/V, Y143R/C/H, Q148H/R/K y N155H) al momento basal, pero mostraban evidencia histórica genotípica de sustituciones a la resistencia INSTI, evidencia fenotípica de resistencia a elvitegravir o raltegravir o evidencia genotípica de sustituciones a la resistencia INSTI en el screening. Las tasas de respuesta en base a genotipo basal se analizaron en un análisis "como tratado" a la semana 48 (n = 175) (Tabla 10). La tasa de respuesta a la semana 48 para los regímenes que contenían dolutegravir fue del 47 % (24 de 51) cuando sustituciones Q148 estaban presentes al inicio del estudio; Q148 siempre estuvo presente con sustituciones a la resistencia INSTI adicionales (ver tabla 10). Además, una respuesta virológica disminuida de un 40% (6 de 15) fue observada cuando la sustitución a E157Q o K se presentó de forma basal con otras sustituciones a la resistencia INSTI pero sin sustituciones Q148H o R.
Respuesta en base a fenotipo basal: Las tasas de respuesta en base a fenotipo basal se analizaron mediante un subgrupo de sujetos que habían alcanzado la semana 24, así como los que descontinuaron o tuvieron rebote antes de la semana 24 (n = 120) (Tabla 11). Estos grupos fenotípicos de inicio se basan en los sujetos ingresados en VIKING-3 y no se supone que representan los puntos de corte definitivos por sensibilidad clínica a dolutegravir. Los datos se proporcionan para guiar a los médicos en la probabilidad de éxito virológico en base a la susceptibilidad a dolutegravir pretratamiento en pacientes resistentes previamente a los INI.
Resistencia a los INSTI emergente al tratamiento: Hubo 50 sujetos con fracaso virológico en el régimen de dolutegravir 2 veces al día en VIKING-3 con el ARN de HIV-1 mayor a 400 copias por ml al momento de fracaso, semana 48 o más, o al momento de la última visita. 39 sujetos con fracaso virológico presentaron datos de resistencia que fueron utilizados en la semana 48. En el análisis de resistencia de la semana 48 un 85% (33 de 39) de los sujetos con fracaso virológico tuvo sustituciones de resistencia INSTI emergentes al tratamiento en sus aislamientos. La sustitución de resistencia INSTI más común emergente al tratamiento fue T97A. Otras sustituciones de resistencia INSTI emergentes frecuentes incluyeron L74M, I o V, E138K o A, G140S, Q148H, R o K, M154I o N155H. Las sustituciones E92Q, Y143R o C/H, S147G, V151A y E157E/Q surgieron en los aislamientos de 1 a 3 de los sujetos. Al fracasar, el factor de cambio medio de dolutegravir desde la referencia fue 61 veces más (rango: 0.75 a 209) para los aislados con sustituciones de resistencia INSTI emergentes (n = 33). La resistencia a uno o más fármacos de régimen de fondo con el régimen de dolutegravir 2 veces al día surgió además en 49% (19 de 39) de los sujetos en el análisis de resistencia a la semana 48. Resistencia cruzada: Cepas mutantes HIV-1 y HIV-2 con resistencia sitio-dirigidas a los INSTI: La susceptibilidad de dolutegravir se probó frente a 60 virus mutantes de sitio dirigido con resistencia INSTI del HIV-1 (28 con una única sustitución y 32 con 2 o más sustituciones) y con 6 virus mutantes de sitio dirigido con resistencia INSTI del HIV-2. Las sustituciones de resistencia individuales a INSTI T66K, I151L, y S153Y confirieron una disminución >2 veces en la susceptibilidad a dolutegravir (rango: 2.3 veces a 3.6 veces de la referencia). Las combinaciones de múltiples sustituciones T66K/L74M, E92Q/N155H, G140C/Q148R, G140S/Q148H, R o K, Q148R/N155H, T97A/G140S/Q148, y sustituciones en E138/G140/Q148 mostraron una disminución >2 veces en la susceptibilidad a dolutegravir (rango: 2.5 veces a 21 veces en relación a la referencia). Para los virus HIV-2 mutantes, las combinaciones de sustituciones 153G/N155H/S163G y E92Q/T97A/N155H/S163D confirieron una disminución de >4 veces en la susceptibilidad a dolutegravir y E92Q/N155H y G140S/Q148R mostraron una disminución de >8.5 veces y >17 veces en susceptibilidad a dolutegravir, respectivamente. Cepas resistentes a los inhibidores de la transcriptasa reversa y a los inhibidores de la proteasa: Dolutegravir demostró actividad antiviral equivalente contra 2 clones mutantes resistentes a INNTR, 3 clones mutantes resistentes a INTR, y 2 clones mutantes resistentes a IP en comparación con la cepa de tipo salvaje. Toxicología no clínica: Carcinogénesis, mutagénesis, reducción de la fertilidad: Carcinogénesis: Estudios de 2 ańos de carcinogenicidad en ratas y ratones se realizaron con dolutegravir. A los ratones se les administró dosis de hasta 500 mg/kg, y a las ratas se les administró dosis de hasta 50 mg/kg. En los ratones no se observaron aumentos significativos en la incidencia de neoplasias relacionadas con los fármacos, a las dosis más altas probadas, resultando en exposiciones AUC a dolutegravir aproximadamente 14 veces más altas que las de los humanos a la dosis recomendada de 50 mg 2 veces al día. En ratas, no se observaron aumentos significativos en la incidencia de neoplasias relacionadas con los fármacos, a las dosis más altas probadas, resultando en exposiciones AUC a dolutegravir 10 veces y 15 veces más alta en machos y hembras, respectivamente, que en las de los humanos a la dosis recomendada de 50 mg 2 veces al día. Mutagénesis: Dolutegravir no fue genotóxico en el ensayo de mutación reversa bacteriano, ni en el ensayo de linfoma de ratón ni en el ensayo de micronúcleos in vivo en roedores. Deterioro de la fertilidad: En un estudio realizado en ratas, no hubo efectos sobre el apareamiento o la fertilidad con dolutegravir hasta 1000 mg/kg/día. Esta dosis se asocia con una exposición que es aproximadamente 24 veces superior a la exposición en los seres humanos a la dosis recomendada de 50 mg 2 veces al día. Estudios clínicos: La eficacia de Tivicay se basa en el análisis de datos de 2 estudios, SPRING-2 (ING113086), SINGLE (ING114467) y FLAMINGO (ING 114915) en sujetos sin tratamiento previo infectados por VIH-1 (n=1641); un estudio, SAILING (ING111762) en sujetos con tratamiento previo sin INSTI (INSTI naďve) infectados por VIH-1 (n=715); y de VIKING-3 (ING112574) en sujetos con tratamiento previo con INSTI infectados por VIH-1 (n=183). El uso de Tivicay en pacientes pediátricos desde los 12 ańos se basa en la evaluación de la seguridad, farmacocinética y eficacia a través de 24 semanas en un ensayo multicéntrico, abierto en sujetos sin resistencia a los INSTI (n=23). Sujetos adultos: Sujetos sin tratamiento previo: En SPRING-2, 822 sujetos fueron aleatorizados y recibieron al menos 1 dosis, ya sea de TIVICAY 50 mg 1 vez al día, o raltegravir 400 mg 2 veces al día, ambos en combinación con tratamiento INTR dual de dosis fija (ya sea sulfato de abacavir y lamivudina [KIVEXA] o emtricitabina / tenofovir [TRUVADA ®]). Hubo 808 sujetos incluidos en los análisis de eficacia y seguridad. Al inicio del estudio, la mediana de edad de los sujetos fue de 36 ańos, 13% mujeres, 15% no blancos, 11% con coinfección por el virus hepatitis B y/o C virus, 2% Clase C del CDC (SIDA), y 28% con ARN de VIH-1 mayor a 100.000 copias por ml, 48% con recuento de linfocitos CD4+ menor a 350 células por mm3, y 39% recibiendo KIVEXA; estas características fueron similares entre los grupos de tratamiento. En SINGLE, 833 pacientes fueron aleatorizados y recibieron al menos 1 dosis, ya sea de Tivicay 50 mg 1 vez al día con una dosis fija de sulfato de abacavir y lamivudina (Kivexa), o una dosis fija de efavirenz/emtricitabina/tenofovir (Atripla®). Al inicio del estudio, la mediana de edad de los sujetos era de 35 ańos, mujeres 16%, el 32% no blanco, el 7% coinfectados por hepatitis C (la coinfección con hepatitis B fue excluida), el 4% Clase C del CDC (SIDA), el 32% con ARN de VIH-1 > 100000 copias/ml, y 53% tenía recuento de linfocitos CD4+ <350 células/mm3, estas características fueron similares entre los grupos de tratamiento. Los resultados a la Semana 96 para SPRING-2 y SINGLE se proporcionan en la Tabla 12. Se utilizó tabulación lateral para simplificar la presentación; comparaciones directas entre los ensayos no se deben hacer debido a que el diseńo de cada estudio es diferente.
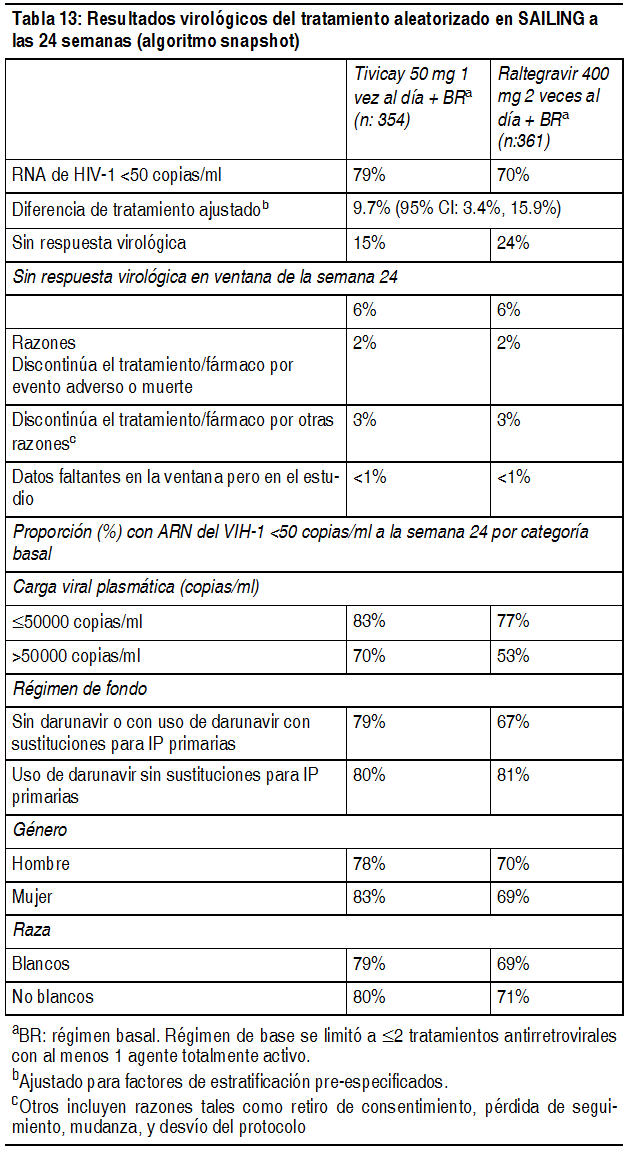
En el estudio SPRING-2, los resultados virológicos también fueron comparables en sus características basales incluyendo: recuento de células CD4+, edad, uso de KIVEXA o TRUVADA® como tratamiento INTR de fondo. El cambio medio en el recuento de células CD4+ desde la basal fue 276 células por mm3 en el grupo recibiendo TIVICAY y 264 células por mm3 para el grupo recibiendo raltegravir a las 96 semanas. No hubo resistencia de tratamiento emergente para dolutegravir o para el NRTI usado como régimen de base. En el estudio SINGLE, se mantuvieron diferencias en tratamiento a través de las características basales incluyendo recuento de células CD4+, la edad, género y raza. Los cambios medios ajustados en los recuentos de células CD4 + desde el inicio fueron de 325 células por mm3 en el grupo que recibió TIVICAY + KIVEXA y 281 células por mm3 en el grupo de ATRIPLA a las 96 semanas. La diferencia ajustada entre los grupos de tratamiento y los IC del 95% fue de 44.0 células por mm3 (14.3 células por mm3, 73.6 células por mm3) (ajustado por factores de estratificación preespecificados: ARN del VIH-1 inicial, recuentos de células CD4+ basales, y la multiplicidad). No hubo resistencia a tratamiento emergente a dolutegravir, abacavir o lamivudina. FLAMINGO: En Flamingo, 485 sujetos fueron aleatorizados y recibieron una dosis ya sea de TIVICAY 50 mg 1 vez al día (n = 243) o de darunavir + ritonavir 800 mg/100 mg 1 vez al día (n = 242), ambos administrados en combinación con un NRTI seleccionado por el investigador como régimen de base (ya sea KIVEXA o TRUVADA). Hubo 484 sujetos incluidos en el análisis de seguridad y eficacia. En la basal, la edad promedio de los sujetos fue de 34 ańos, 15% mujeres, 28% no blancos, 10% tenía hepatitis B y/o coinfección con virus hepatitis C, 3% fueron considerados Clase C de la CDC, 25% tenía VIH-1 RNA mayor a 100.000 copias por ml, y 3% tenía conteo de células CD4+ menor a 350 células por ml; estas características fueron similares entre los grupos de tratamiento. La tasa general de respuesta por algoritmo Snapshot en la semana 48 fue de un 90% para TIVICAY y 83% para darunavir/ritonavir. La diferencia ajustada a proporcional e IC 95% fue de 7.1% (0.9; 13.2). No se observó aparición de resistencia primaria al tratamiento en ninguno de los grupos en tratamiento. Con tratamiento previo pero sin inhibidores de transferencia de hebra (INSTI-naďve): En el ensayo internacional, multicéntrico, doble ciego (SAILING), 719 adultos infectados por el VIH-1 con tratamiento previo con antirretrovirales fueron aleatorizados y recibieron ya sea Tivicay 50 mg 1 vez al día o raltegravir 400 mg 2 veces al día con régimen de base seleccionados por el investigador con un máximo de 2 agentes, incluyendo al menos 1 agente completamente activo. Hubo 715 sujetos incluidos en los análisis de eficacia y seguridad. Al inicio del ensayo, la mediana de edad fue de 43 ańos, 32% mujeres, 50% no blancos, 16% coinfectados con el virus de la hepatitis B y/o C, 46% Clase C del CDC (SIDA), 20% con ARN del VIH-1 > 100000 copias/ml, y 72% con recuento de células CD4+ <350 células/mm3; estas características fueron similares entre los grupos de tratamiento. Todos los sujetos tenían al menos resistencia a 2 clases de antirretrovirales, y el 49% de los sujetos tenían al menos resistencia a 3 clases de antirretrovirales al inicio del estudio. Los resultados de SAILING a la Semana 48 se muestran en la Tabla 13.
Se mantuvieron diferencias en tratamiento a través de las características basales incluyendo recuento de células CD4+ y la edad. Los cambios promedio ajustados en los recuentos de células CD4 + desde el basal fueron de 114 células/mm3 en el grupo que recibió Tivicay y 106 células/mm3 en el grupo de raltegravir. Sujetos con tratamiento previo con inhibidores de transferencia de hebra (INSTI): El ensayo VIKING-3 examinó el efecto de Tivicay 50 mg 2 veces al día durante 7 días de monoterapia funcional, seguido por un régimen de base optimizado y tratamiento continuado de Tivicay 50 mg 2 veces al día. En este ensayo multicéntrico, abierto, de un solo brazo VIKING-3, 183 adultos infectados por VIH-1 con tratamiento previo con antirretrovirales con fracaso virológico y evidencia actual o histórica de resistencia a raltegravir y/o elvitegravir se les administró Tivicay 50 mg 2 veces al día con el régimen de base que había fracasado por 7 días. Seguido por la administración de Tivicay con un OBT a partir del día 8. Se ingresaron un total de 183 sujetos: 133 sujetos con resistencia INSTI en el screening y 50 sujetos con evidencia solamente histórica de resistencia (no en el screening). Al inicio, la mediana de edad de los pacientes era de 48 ańos, 23% eran mujeres, 29% no eran blancos, y 20% tenían coinfección por hepatitis B y/o C. La mediana basal de recuento de CD4+ fue de 140 células/mm3, la mediana de la duración del TAR previo fue de 13 ańos, y 56% eran Clase C del CDC. Los sujetos mostraron resistencia a múltiples clases de antirretrovirales en la basal: 79% tenían ≥2 mutaciones para NRTI, 75% ≥1 para NNRTI, y 71% ≥2 para inhibidores de la proteasa (IP); 62% tenían virus no R5. El cambio promedio de reducción desde la basal en el ARN del VIH-1 al día 8 (criterio de valoración primario) fue de 1.4 log10 (IC del 95% 1.3 - 1.5 log10, p<0.001). La respuesta a la semana 48 se vio afectada por sustituciones INSTI basales (véase Microbiología). Después de la fase de monoterapia funcional, los sujetos tuvieron la oportunidad de optimizar su régimen de base, siempre que fuera posible. Resultados virológicos a la semana 48 para VIKING-3 se muestran en la Tabla 14.
Sujetos albergando el virus con sustitución Q148 y con sustituciones secundarias adicionales asociados a Q148 también tuvieron una respuesta reducida de manera escalonada a la semana 48 (véase Microbiología). El cambio promedio en el recuento de células CD4 + desde el inicio fue de 65 células/mm3 a la semana 48. Sujetos pediátricos: IMPAACT P1093 es un ensayo de fase 1/2, de 48 semanas, multicéntrico, abierto para evaluar los parámetros farmacocinéticos, la seguridad, tolerabilidad y eficacia de Tivicay en los regímenes de tratamiento de combinación en bebés, nińos, y adolescentes infectados por el HIV-1. La etapa inicial de búsqueda de dosis incluyó la evaluación intensiva farmacocinética en 10 sujetos sin tratamiento previo con INSTI (INSTI-naďve) (de 12 a 18 ańos de edad). La selección de la dosis se basó en lograr una exposición plasmática y una concentración estacionaria de dolutegravir semejante a la de los adultos. Después de seleccionar la dosis, 13 sujetos adicionales fueron ingresados para evaluar la seguridad a largo plazo, la tolerabilidad y la eficacia. Estos 23 sujetos tenían una mediana de edad de 14 ańos (rango: 12 a 17), eran 78% nińas y 52% negros. Al inicio del estudio, la media plasmática de ARN del VIH-1 fue de 4.3 log10 copias/ml, la media de recuento de CD4+ fue de 466 células/mm3 (rango: 11 a 1025), y la media de CD4 +% fue del 22% (rango de 1% a 39%). En general, el 17% tenía nivel plasmático basal de ARN del VIH-1 > 50000 copias/ml y el 39% eran Clase C del CDC. La mayoría de los sujetos habían utilizado previamente, al menos, 1 NNRTI (52%) o 1 IP (78%). A las 24 semanas, el 70% de los sujetos tratados con Tivicay 1 vez al día (35 mg: n=4, 50 mg: n=19), más terapia de base optimizada logró una carga viral de <50 copias/ml. La media de aumento de recuento de células CD4+ (porcentaje) desde el inicio hasta la semana 24 fue de 63 células/mm3 (5%).
Posología: Los comprimidos de Tivicay se pueden tomar con o sin alimentos. Adultos: Tabla 1: Dosificación recomendaciones para Tivicay en pacientes adultos: Población: Dosis recomendada: Sin tratamiento previo (naďve) o con tratamiento previo sin inhibidores de transferencia de hebra de la integrasa (INSTI naďve): 50 mg 1 vez al día. Sin tratamiento previo (naďve) o con tratamiento previo sin inhibidores de la transferencia de hebra de la integrasa (INSTI naďve) cuando es coadministrado con los siguientes inductores potentes de los UGT1A/CYP3A: efavirenz, fosamprenavir/ritonavir, tipranavir/ritonavir, o rifampicina: 50 mg 2 veces al día. Con tratamiento previo con inhibidores de la transferencia de hebra (INSTI) y con ciertas sustituciones de resistencia a los INSTI asociadas o una sospecha clínica de resistencia a los INSTI* [véase Microbiología]: 50 mg 2 veces al día. * Se deben considerar combinaciones alternativas que no incluyen inductores metabólicos siempre que sea posible (véase Interacciones). No se han evaluado la seguridad y la eficacia de dosis superiores a 50 mg 2 veces al día. Pacientes pediátricos: Sin tratamiento previo (naďve) o con tratamiento previo sin inhibidores de transferencia de hebra de la integrasa (INSTI naďve): La dosis recomendada en pacientes pediátricos mayores de 12 ańos con un peso mínimo de 40 kg es de 50 mg administrados 1 vez al día. Si efavirenz, fosamprenavir/ritonavir, tipranavir/ritonavir, o rifampicina se coadministran, la dosis recomendada de Tivicay es de 50 mg 2 veces al día. La seguridad y la eficacia de Tivicay no se han establecido en pacientes pediátricos menores de 12 ańos o con un peso menor a 40 kg, o en pacientes pediátricos con tratamiento INSTI previo con resistencia, sea documentada o sospechada clínicamente a otros INSTI (raltegravir, elvitegravir).
Modo de Empleo:Instrucciones para el uso/manejo: No existen requisitos especiales para el uso o manejo de este producto. No están disponibles todas las presentaciones en todos los países.
Efectos Colaterales:Las siguientes reacciones adversas a los medicamentos (eventos adversos evaluados de estar causalmente relacionados por el investigador o reacciones farmacológicas adversas [ADR en inglés]) se tratan en otras secciones del folleto: Las reacciones de hipersensibilidad (véase Advertencias y Precauciones). Efectos sobre la bioquímica hepática en suero en pacientes coinfectados por hepatitis B o C (véase Advertencias). Redistribución grasa (véase Advertencias y Precauciones). Síndrome de reconstitución inmune (véase Advertencias y Precauciones). Dado que los ensayos clínicos se llevan a cabo en condiciones muy variables, las tasas de reacciones adversas observadas en los ensayos clínicos de un fármaco no pueden compararse directamente con las tasas en los ensayos clínicos de otro fármaco y pueden no reflejar las tasas observadas en la práctica. Experiencia con ensayos clínicos en sujetos adultos: Reacciones adversas emergentes al tratamiento (ADR): Sujetos sin tratamiento previo (naďve): Esta evaluación de seguridad de Tivicay en sujetos infectados por HIV-1 sin tratamiento previo se basa en el análisis de datos a las 48 semanas de 2 ensayos en curso, doble-ciegos, internacionales multicéntricos. SPRING-2 (ING113086) y SINGLE (ING114467) y datos a las 48 semanas del estudio internacional, multicéntrico, abierto FLAMINGO (ING114915). En SPRING-2, 822 pacientes fueron aleatorizados y recibieron al menos 1 dosis de Tivicay 50 mg 1 vez al día o 1 dosis de raltegravir 400 mg 2 veces al día, ambas en combinación con tratamiento de inhibidores nucleótidos de la transcriptasa reversa (NRTI) con dosis fija (ya sea sulfato de abacavir y lamivudina [Kivexa®] o emtricitabina/tenofovir [Truvada®]). Hubo 808 sujetos incluidos en los análisis de eficacia y seguridad. La tasa de eventos adversos que llevaron a la interrupción del tratamiento fue de 2% en ambos brazos. En SINGLE, 833 pacientes fueron aleatorizados y recibieron al menos 1 dosis ya sea de Tivicay 50 mg con sulfato de abacavir y lamivudina (Kivexa®) de dosis fija 1 vez al día o efavirenz/emtricitabina/tenofovir (Atripla®) de dosis fija 1 vez al día. La tasa de eventos adversos que condujeron a la discontinuación fue de 2% en los sujetos que recibieron Tivicay 50 mg 1 vez al día + Kivexa y 10% en los sujetos que recibieron Atripla 1 vez al día. Reacciones adversas a medicamento (RAMS) de moderada a severa intensidad emergentes al tratamiento observadas en al menos un 2% de los sujetos de cualquiera de los brazos de tratamientos en los estudios SPRING-2 y SINGLE, se proporcionan en la Tabla 2. Se utilizó tabulación lateral para simplificar la presentación; comparaciones directas entre los estudios no se deben hacer debido a que el diseńo de cada estudio es diferente.
Además, el insomnio Grado 1 se informó en un 1% y menos de un 1% de los sujetos recibiendo Tivicay y raltegravir, respectivamente, en SPRING-2, mientras que en SINGLE las tasas fueron del 7% y 3% para Tivicay y Atripla, respectivamente. Estos eventos no limitaron el tratamiento. En un estudio abierto, multicéntrico (FLAMINGO), 243 sujetos recibieron TIVICAY 50 mg 1 vez al día versus 242 sujetos quienes recibieron darunavir 800 mg/ritonavir 100 mg 1 vez al día, ambos en combinación con un régimen de base de NRTI seleccionado por el investigador (KIVEXA o bien TRUVADA). Hubo 484 sujetos incluidos en el análisis de seguridad y eficacia. A través de 48 semanas, las tasas de eventos adversos que llevaron a discontinuación fueron de un 2% en sujetos recibiendo TIVICAY y 4% en sujetos recibiendo darunavir/ritonavir. Las reacciones adversas observadas en FLAMINGO fueron generalmente consistentes con aquellas vistas en SPRING-2 y SINGLE. Sujetos con tratamiento previo sin inhibidores de integrasa (INSTI naďve): En un estudio doble ciego internacional multicéntrico (ING111762, SAILING), 719 adultos infectados por HIV-1 con tratamiento previo con antirretrovirales fueron aleatorizados y recibieron ya sea Tivicay 50 mg 1 vez al día o raltegravir 400 mg 2 veces al día con regímenes de fondo seleccionados por los investigadores con un máximo de 2 agentes, incluyendo al menos 1 agente completamente activo. A las 48 semanas, las tasas de eventos adversos que provocaron una interrupción fueron de 3% en los sujetos que recibieron Tivicay 50 mg 1 vez al día + régimen de fondo y 4% en los sujetos que recibieron raltegravir 400 mg 2 veces al día + régimen de fondo. La única reacción farmacológica adversa (RAM) emergente al tratamiento de intensidad moderada a severa con frecuencia de al menos ≥2% en cualquiera de los brazos de tratamiento fue la diarrea, 2% (5/354) en los sujetos recibiendo Tivicay 50 mg 1 vez al día + régimen de fondo y 2% (65/361) en los sujetos recibiendo raltegravir 400 mg 2 veces al día + régimen de fondo. Sujetos con tratamiento previo con inhibidores de integrasa (INSTI): En un ensayo multicéntrico, abierto, de un solo brazo (ING112574, VIKING-3), 183 adultos infectados por HIV-1 tratados previamente con antirretrovirales con fracaso virológico y con evidencia actual o histórica de resistencia a raltegravir y/o elvitegravir recibieron Tivicay 50 mg 2 veces al día con el actual régimen de fondo fallido por 7 días seguido por un tratamiento de base optimizado empezando el 8ş día. La tasa de eventos adversos que condujeron a la descontinuación fue de 4% de los sujetos a la semana 48. Las reacciones farmacológicas adversas (ADRs) emergentes al l tratamiento en VIKING-3 fueron generalmente similares en comparación con las observadas con la dosis de 50 mg 1 vez al día en los estudios de Fase 3 en adultos. Reacciones adversas menos comunes observadas en los ensayos con sujetos sin tratamiento previo (naďve) y con tratamiento previo: Las siguientes reacciones adversas ocurrieron en menos de un 2% de los sujetos sin tratamiento previo o con tratamiento previo recibiendo Tivicay en un régimen combinado en cualquiera de los estudios. Estos eventos se han incluido por su gravedad y la evaluación de una posible relación causal. Trastornos gastrointestinales: Dolor abdominal, malestar abdominal, flatulencia, dolor abdominal superior, vómitos. Trastornos generales: Fatiga. Trastornos hepatobiliares: Hepatitis. Trastornos musculoesqueléticos: Miositis. Trastornos psiquiátricos: Ideación, intentos, comportamientos suicidas o suicidio. Estos eventos se observaron principalmente en sujetos con historial pre-existente de depresión u otra enfermedad psiquiátrica. Trastornos renales y urinarios: Insuficiencia renal. Trastornos de la piel y del tejido subcutáneo: Prurito. Anormalidades de laboratorio: Sujetos sin tratamiento previo (naďve): Anormalidades de laboratorio seleccionadas (Grados 2 a 4) con un grado de deterioro con respecto a la línea base y representando el peor grado de toxicidad en ≥2% de los sujetos se presentan en la Tabla 3. El cambio medio con respecto a la línea base observada en los valores de lípidos seleccionados se presenta en la Tabla 4. Se utilizó tabulación lateral para simplificar la presentación; comparaciones directas entre los ensayos no se deben hacer debido a que el diseńo de cada ensayo es diferente.
Las anomalidades de laboratorio observadas en el estudio FLAMINGO fueron generalmente consistentes con las observaciones en SPRING-2 y SINGLE. Sujetos con tratamiento previo sin inhibidores de integrasa (INSTI naďve): Las anormalidades de laboratorio observadas en SAILING fueron generalmente similares en comparación con las observaciones vistas en los estudios de sujetos sin tratamiento previo (SPRING -2 y SINGLE). Sujetos con tratamiento previo, con inhibidores de transferencia de hebra de la integrasa (INSTI): Las anormalidades de laboratorio emergentes del tratamiento más frecuentes (mayores que 5% para los grados 2 a 4 combinado) observadas en VIKING-3 a la semana 48 fueron elevaciones de ALT (9%), AST (8%), colesterol (10%), cretinquinasa (6%), hiperglucemia (14%) y lipasa (10%). El 2% (4 de 183) de los sujetos tenían una anomalía de hematología grado 3 y 4 y emergente al tratamiento, donde la neutropenia (2% [3 de 183]) fue la más frecuentemente reportada. Coinfección por hepatitis B y/o C: En los ensayos de Fase 3, los sujetos coinfectados por hepatitis B y/o C se les permitió ingresar siempre y cuando sus niveles basales de pruebas de función hepática no fueran más de 5 veces el límite superior del rango de referencia normal. En general, el perfil de seguridad en sujetos coinfectados por hepatitis B y/o C fue similar al observado en sujetos sin coinfección por hepatitis B o C, aunque las tasas de anomalías de AST y ALT fueron más altas en el subgrupo de sujetos coinfectados con hepatitis B y/o C de todos los brazos de tratamiento. Anomalías de ALT de grados 2 a 4 en coinfectados con hepatitis B y/o C en comparación con sujetos monoinfectados con HIV-1 recibiendo Tivicay fueron observados en el 16% vs 2%, con la dosis de 50 mg 1 vez al día y el 8% vs 7% con la dosis de 50 mg 2 veces al día. Pruebas de función hepática elevadas consistentes con el síndrome de reconstitución inmune se observaron en algunos sujetos con hepatitis B y/o C al inicio de la terapia con Tivicay, en particular en los casos en los que la terapia antihepatitis fue retirada (véase Advertencias). Cambios en la creatinina sérica: Se ha demostrado que dolutegravir aumenta la creatinina sérica, debido a la inhibición de la secreción tubular de creatinina sin que afecte la función renal glomerular (véase Farmacología clínica). Aumentos de la creatinina sérica ocurrieron durante las primeras 4 semanas de tratamiento y se mantuvieron estables a través de las 48 a 96 semanas. En sujetos sin tratamiento previo, un cambio medio con respecto al nivel basal de 0.11 mg/dl (rango: -0.60 mg/dl a 0.65 mg/dl) se observó después de 96 semanas de tratamiento. Incrementos de creatinina fueron comparables independiente de los inhibidores nucleotídicos de la transcriptasa inversa (NRTI) usados como terapia de base y fueron similares en pacientes que habían sido tratados previamente. Experiencia con estudios clínicos en sujetos pediátricos: IMPAACT P1093 es un estudio en curso, multicéntrico, abierto, no comparativo de aproximadamente 160 sujetos pediátricos infectados con HIV-1 entre 6 meses y 18 ańos de edad, de los cuales 23 sujetos ingresados tuvieron tratamiento previo sin inhibidores de la transferencia de hebra de la integrasa (INSTI naďve) con edades entre 12 ańos y 18 ańos de edad (véase Uso en poblaciones especiales, estudios clínicos). El perfil de reacciones adversas fue similar al de los adultos. Reacciones de Grado 2 reportados en al menos 1 sujeto fueron erupción (n= 1), dolor abdominal (n= 1) y diarrea (n= 1). No se reportaron reacciones adversas de grado 3 a 4. Las anomalías de laboratorio de grado 3 fueron bilirrubina total elevada y lipasa aumentada, reportados en 1 sujeto cada una. No se reportaron anomalías de laboratorio de grado 4. Los cambios en la creatinina sérica media fueron similares a los observados en adultos.
Contraindicaciones:Tivicay está contraindicado en pacientes: Con reacción de hipersensibilidad previa a dolutegravir: Que reciben dofetilida, debido a la posibilidad de aumento de las concentraciones plasmáticas de dofetilida y el riesgo de eventos graves y/o potencialmente mortales (véase Interacciones farmacológicas).
Advertencias:Reacciones de hipersensibilidad: Se han reportado reacciones de hipersensibilidad con inhibidores de integrasa, incluyendo Tivicay, las cuales se caracterizaron por erupción cutánea, hallazgos constitucionales, y en ocasiones en disfunción orgánica, incluyendo dańo hepático. Los eventos fueron reportados en menos de un 1% de los sujetos que recibieron Tivicay en ensayos clínicos de Fase 3. Suspenda Tivicay y otros agentes sospechosos inmediatamente si se presentan signos o síntomas de reacciones de hipersensibilidad (incluyendo, pero no limitado a erupción grave o erupción acompańada de fiebre, malestar general, fatiga, mialgias o artralgias, ampollas o descamación de la piel, ampollas orales o lesiones, conjuntivitis, edema facial, hepatitis, eosinofilia, angioedema, dificultad para respirar). Debe monitorearse el estado clínico, incluyendo las aminotransferasas hepáticas, así como iniciar el tratamiento adecuado. El retraso en la interrupción del tratamiento con Tivicay u otros agentes sospechosos después de la aparición de hipersensibilidad, puede causar una reacción potencialmente mortal. Tivicay está contraindicado en pacientes que hayan experimentado una reacción de hipersensibilidad previa a Tivicay. Efectos sobre la prueba de bioquímica hepática en pacientes coinfectados por hepatitis B o C: Pacientes con hepatitis B o C subyacente pueden estar en mayor riesgo de empeoramiento o desarrollo de elevaciones de las transaminasas con el uso de Tivicay (véase Efectos colaterales). En algunos casos, las elevaciones de las transaminasas fueron consistentes con el síndrome de reconstitución inmunitaria o reactivación de hepatitis B, en particular en situaciones en las que se retiró la terapia antihepatitis. Se recomienda el monitoreo de las pruebas de función hepática antes y durante el tratamiento con Tivicay en pacientes con enfermedad hepática subyacente, como la hepatitis B o C. Redistribución grasa: La redistribución/acumulación de la grasa corporal, incluyendo obesidad central, aumento de la grasa dorsocervical (joroba de búfalo), emaciación periférica, emaciación facial, agrandamiento de las mamas y cara de luna llena, se han observado en pacientes que reciben terapia antirretroviral. El mecanismo y las consecuencias a largo plazo de estos eventos no se conocen actualmente. No se ha establecido una relación causal. Síndrome de reconstitución inmune: El síndrome de reconstitución inmune se ha reportado en pacientes tratados con terapia antirretroviral combinada, incluyendo Tivicay. Durante la fase inicial del tratamiento antirretroviral combinado, los pacientes cuyos sistemas inmunes responden, pueden desarrollar una respuesta de inflamación frente a infecciones oportunistas indolentes o residuales (tales como infección por Mycobacterium avium, citomegalovirus, neumonía por Pneumocystis jirovecii [PCP], o tuberculosis), que puede requerir una evaluación adicional y tratamiento. Trastornos autoinmunes (tales como la enfermedad de Graves, polimiositis, y el síndrome de Guillain-Barré) también se han reportado durante la reconstitución inmune, sin embargo, el tiempo de inicio es más variable y puede ocurrir meses después del inicio del tratamiento.
Precauciones:Uso en poblaciones especiales: Embarazo: Embarazo - categoría B. No existen estudios adecuados y bien controlados en mujeres embarazadas. Dado que los estudios de reproducción en animales no siempre son predictivos de la respuesta humana, y se demostró que dolutegravir cruza la placenta en estudios con animales, este medicamento debe utilizarse durante el embarazo sólo si es claramente necesario. Datos en animales: Los estudios de reproducción se realizaron en ratas y conejos a dosis de hasta 27 veces la dosis humana de 50 mg 2 veces al día y no revelaron evidencia de alteración de la fertilidad o dańo al feto debido a Tivicay. La administración oral de dolutegravir a ratas preńadas a dosis de hasta 1000 mg/kg al día, aproximadamente 27 veces la exposición humana clínica de 50 mg 2 veces al día basada en el AUC, desde el día 6 al 17 de gestación no provocó toxicidad materna, toxicidad en el desarrollo, o teratogenicidad. La administración oral de dolutegravir a conejas preńadas a dosis de hasta 1000 mg/kg al día, aproximadamente 0.4 veces la exposición clínica en humanos de 50 mg 2 veces al día en base al AUC, desde el día 6 al 18 de gestación, no provocó toxicidad en el desarrollo o teratogenicidad. En conejos, la toxicidad materna (disminución del consumo de alimentos, escasa/nada de heces/orina, supresión en el aumento de peso corporal) se observó a 1000 mg/kg. Madres lactantes: Los centros para el control y la prevención de enfermedades recomiendan que las madres infectadas por el HIV-1 en los Estados Unidos no amamanten a sus hijos para evitar arriesgarse a la transmisión postnatal de la infección por HIV-1. Los estudios en ratas lactantes y sus crías indican que dolutegravir estuvo presente en la leche de las ratas. No se sabe si dolutegravir se excreta en la leche humana. Debido tanto a la posibilidad de transmisión del HIV y la posibilidad de reacciones adversas en los lactantes, instruir a las madres acerca de no amamantar. Uso pediátrico: La seguridad y la eficacia de Tivicay no se han establecido en pacientes pediátricos menores de 12 ańos pesando menos de 40 kg en cualquier paciente pediátrico con tratamiento INSTI previo con resistencia, sea documentada o sospechada clínicamente, a otros INSTI (raltegravir, elvitegravir). La seguridad, las respuestas virológica e inmunológica en sujetos que recibieron Tivicay se evaluó en 23 pacientes infectados por el HIV-1 con tratamiento previo sin inhibidores de la transferencia de hebra (INSTI-naďve) mayores o igual a 12 ańos y menores a 18 ańos del ensayo clínico abierto, multicéntrico, de búsqueda de dosis IMPAACT P1093 (véase Efectos colaterales, Farmacología clínica, Estudios clínicos). Los parámetros farmacocinéticos evaluados en 9 pacientes con peso de al menos 40 kg que recibieron 50 mg diarios y 1 sujeto (con peso 37 kg) que recibió 35 mg 1 vez al día, fueron similares a los de adultos que recibieron 50 mg 1 vez al día. Véase Posología para consultar las recomendaciones de dosificación para pacientes pediátricos de 12 ańos o más y con un peso mínimo de 40 kg. Frecuencia, tipo y gravedad de las reacciones adversas a los medicamentos en pacientes pediátricos fueron similares a los observados en adultos (véase Efectos colaterales). Uso geriátrico: Los ensayos clínicos de Tivicay no incluyeron un número suficiente de sujetos de 65 ańos y mayores para determinar si responden de manera diferente a los sujetos más jóvenes. En general, se debe tener precaución al administrar Tivicay en pacientes de edad avanzada dado a la mayor frecuencia de disminución de las funciones hepática, renal o cardíaca, y de enfermedades concomitantes u otra terapia farmacológica (véase Farmacología clínica). Insuficiencia hepática: No se observaron diferencias farmacocinéticas clínicamente importantes entre los sujetos con insuficiencia hepática moderada y sujetos sanos control. No es necesario ajustar la dosis en pacientes con insuficiencia hepática leve a moderada (puntuación de Child-Pugh A o B). El efecto de la insuficiencia hepática severa (puntuación Child-Pugh C) sobre la farmacocinética de Dolutegravir no se ha estudiado. Por lo tanto, Tivicay no está recomendado para uso en pacientes con insuficiencia hepática severa (véase Farmacología clínica). Insuficiencia renal: Las concentraciones plasmáticas de dolutegravir se redujeron en los sujetos con insuficiencia renal severa en comparación con los controles sanos. Sin embargo, un ajuste de dosis no es necesario para pacientes sin tratamiento previo o tratamiento previo sin inhibidores de la integrasa (INI naďve) con insuficiencia renal leve, moderada o severa o para pacientes con tratamiento INI previo (con ciertas sustituciones a la resistencia a INI asociadas o sospecha clínica de resistencia a los INI) con insuficiencia renal leve o moderada. Se debe tener precaución en pacientes con tratamiento INI previo (con ciertas sustituciones a la resistencia INI o sospecha clínica de resistencia a INI [véase Microbiología]). Con insuficiencia renal severa, ya que la disminución de las concentraciones de dolutegravir puede resultar en pérdida del efecto terapéutico y el desarrollo de resistencia a Tivicay u otros agentes antirretrovirales coadministrados (véase Farmacología clínica). Dolutegravir no se ha estudiado en pacientes en diálisis.
Interacciones Medicamentosas:Efecto de dolutegravir sobre la farmacocinética de otros agentes: In vitro, dolutegravir inhibió los transportadores de cationes orgánicos renal, OCT2 (IC50 = 1.93 µM) y transportadores de multidrogas y toxinas de extrusión (MATE)1 (CI50 = 6.34 µM). In vivo, dolutegravir inhibe la secreción tubular de creatinina mediante la inhibición de OCT2 y potencialmente de MATE1. Dolutegravir puede aumentar las concentraciones plasmáticas de los fármacos eliminados a través de OCT2 o MATE1 (dofetilida y metformina, Tabla 5) (véase Contraindicaciones, Interacciones farmacológicas). In vitro, dolutegravir inhibió los transportadores renales basolaterales, transportadores de aniones orgánicos (OAT)1 (CI50 = 2.12 µM) y OAT3 (CI50 = 1.97 µM). Sin embargo, in vivo, dolutegravir no alteró las concentraciones plasmáticas de tenofovir o paraaminohipurato, sustratos de OAT1 y OAT3. In vitro, dolutegravir no inhibió (IC50 >50 µM) a: Citocromo P450 (CYP)1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP3A, UGT1A1, UGT2B7, glicoproteína P (P-gp), la proteína de resistencia al cáncer de mama (BCRP), bomba exportadora de sales biliares (BSEP), polipéptido transportador de aniones orgánicos (OATP)1B1, OATP1B3, OCT1, proteína de resistencia a múltiples fármacos (MRP)2 o MRP4. In vitro, dolutegravir no indujo a CYP1A2, CYP2B6 o CYP3A4. En base a estos datos y los resultados de los ensayos de interacción farmacológica, no se espera que dolutegravir afecte la farmacocinética de los fármacos que son sustratos de estas enzimas o transportadores. En los estudios de interacción farmacológica, dolutegravir no tuvo un efecto clínicamente relevante sobre la farmacocinética de los siguientes medicamentos: Tenofovir, metadona, midazolam, rilpivirina y anticonceptivos orales que contienen norgestimato y etinil estradiol. Utilizando comparaciones entre estudios de los datos farmacocinéticos históricos para cada medicamento que interactuó, dolutegravir parece no haber afectado a la farmacocinética de los siguientes medicamentos: atazanavir, darunavir, efavirenz, etravirina, fosamprenavir, lopinavir, ritonavir y telaprevir. Efecto de otros fármacos sobre la farmacocinética de dolutegravir: Dolutegravir es metabolizado por UGT1A1 con alguna contribución de CYP3A. Dolutegravir también es un sustrato de la UGT1A3, UGT1A9, BCRP y P-gp in vitro. Los fármacos que inducen estas enzimas y transportadores pueden disminuir la concentración plasmática de dolutegravir y reducir el efecto terapéutico de dolutegravir. La coadministración de dolutegravir y otros fármacos que inhiben estas enzimas puede aumentar la concentración plasmática de dolutegravir. Etravirina redujo significativamente las concentraciones plasmáticas de dolutegravir, pero el efecto de etravirina fue mitigado por la coadministración de lopinavir/ritonavir o darunavir/ritonavir, y se espera que sea mitigado por atazanavir/ritonavir (Tabla 5) (véase Interacciones farmacólogicas, Farmacología clínica). Darunavir/ritonavir, lopinavir/ritonavir, rilpivirina, tenofovir, boceprevir, telaprevir, prednisona, rifabutina y omeprazol no tuvieron ningún efecto clínicamente significativo sobre la farmacocinética de dolutegravir. Interacciones entre drogas establecidas y otras potencialmente significativas: La tabla 5 proporciona recomendaciones clínicas como resultado de las interacciones farmacológicas con Tivicay. Estas recomendaciones se basan ya sea en ensayos de interacción farmacológica o en interacciones previstas debido a la magnitud esperada de la interacción y las potenciales reacciones adversas graves o pérdida de eficacia (véase Posología, Farmacología clínica).
Sobredosificación: No existe un tratamiento específico para la sobredosis de Tivicay. Si ocurre una sobredosis, el paciente debe ser tratado con un monitoreo adecuado según sea necesario. Debido a que dolutegravir presenta una alta unión a proteínas plasmáticas, es poco probable que se elimine mediante diálisis.
Incompatibilidades: No se han identificado incompatibilidades.
Conservación:Vida de anaquel: La fecha de caducidad se muestra en el envase. Precauciones especiales para el almacenamiento: No almacene a más de 30°C.
Presentaciones:Naturaleza y contenido del envase: Los comprimidos de Tivicay se suministran en frascos de HDPE (polietileno de alta densidad).
 Laboratorio
Laboratorio